

 PDF
PDF Citation
Citation Print
Print



INTRODUCTION
Ovarian cancer (OC) is the main cause of death from gynecologic malignant tumors. In 2012, there were an estimated 238,700 new cases of OC worldwide, which led to more than 150,000 deaths [1]. The high lethality of this cancer is attributed to its limited screening methods and nonspecific symptoms that result in many patients only being diagnosed at an advanced stage (International Federation of Gynecology and Obstetrics [FIGO] stage III and IV). Up to 90% of patients with early stage OC can be cured through surgery and chemotherapy, and the 5-year survival rate after initial diagnosis of early stage OC is more than 70%. However, this rate dramatically decreases to less than 30% for those with advanced-stage OC [23]. Currently, debulking surgery in combination with platinum-taxane-based chemotherapy is the standard and first-line treatment for OC. Although approximately 80% of OC patients go into remission initially, more than 60% relapse within 16–18 months [45]. Therefore, novel complementary methods including immunotherapy, and especially combined immunotherapy, are needed to improve the clinical outcome of OC patients.
Studies have demonstrated that tumor cells can escape a host's immune attack through several pathways that constitute the cancer-immune escape system [67]. The most important pathway in this escape system is an immune checkpoint signal called the programmed death-1 (PD-1) pathway [8]. Evidence suggests that OCs are immunogenic tumors; thus, they should be treatable using immunotherapy [9101112]. In previous studies, employing immunotherapy to block the immune checkpoint (PD-1 pathway) has shown enormous potential and had a remarkable clinical efficacy, prolonging overall survival (OS) and progression-free survival (PFS) in cancer patients with non-small-cell lung cancer, melanoma, and renal cell carcinoma [1314].
A single agent blocking the PD-1 pathway has clearly demonstrated specific antitumor activity in multiple malignant tumor types by reversing the suppression of T cell function and the restriction of tumor cell killing [1516], thus increasing patient survival rate [14]. However, the patients who respond to this treatment are only those with early OC [817]. In advanced tumors, the immune response is inhibited by the suppressive effect of the tumor microenvironment and the multiple immunological tolerance mechanisms (e.g., regulatory T cells [Tregs] and myeloid-derived suppressor cells [MDSCs]) [18]. Based on these findings, scholars proposed that advanced cancer cases may benefit from combined immunotherapies. On September 30, 2015, the Food and Drug Administration (FDA) approved nivolumab (a PD-1-blocking antibody, Opdivo®; Bristol-Myers Squibb, Princeton, NJ, USA) in combination with ipilimumab (a cytotoxic T-lymphocyte-associated protein-4 [CTLA-4]-blocking antibody, Yervoy®; Bristol-Myers Squibb) for the treatment of patients with advanced melanoma. This combined immunotherapy may provide a new direction and proof-of-concept evidence for OC. In addition, several preclinical and clinical studies have shown the therapeutic efficacy of monoclonal antibodies (mAbs) combined with chemotherapy, targeted therapy, radiotherapy, immunotherapy, or other agents for treating OC [192021222324].
In this review, we summarize the most up-to-date data on these combinations for OC and discuss the promising clinical activities and the toxicities observed using such combinations.
PD-1/PD-1 LIGAND SIGNALING
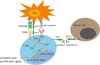
The PD-1 gene was discovered in 1992 as a gene upregulated in a T cell hybridoma and hematopoietic progenitor cell line [25]. PD-1 is a 288-amino acid (aa) type-I transmembrane protein that is composed of an immunoglobulin (Ig) superfamily domain, the stalk of approximately 20-aa, the transmembrane domain, and intracellular domain of approximately 95 residues containing an immunoreceptor tyrosine-based inhibitory motif (ITIM) and an immunoreceptor tyrosine-based switch motif (ITSM). ITSMs are essential for the delivery of inhibitory signals [26]. The C-terminal tyrosine of PD-1 is highly conserved in mice and humans and is related to Src homology region 2 domain-containing phosphatase-1 (SHP-1) and SHP-2. However, N-terminal tyrosine is not related to SHP-1 or SHP-2 [27]. PD-1 ligation, along with T cell receptor (TCR) signaling, leads to phosphorylation of the cytoplasmic tyrosine domain and the recruitment of SHP-2 to the C-terminal tyrosine in an ITSM. Thereafter, SHP-2 dephosphorylates Ras and the phosphatidylinositol 3-kinase (PI3K) pathway, which triggers the inhibition of downstream signaling. Subsequently, the blockade of Akt signaling results in a decrease in cytokine production and T cell proliferation and survival [2829] (Fig. 1). PD-1 is expressed on T cells, B cells, natural killer T (NKT) cells, dendritic cells (DCs), and activated monocytes [8]. The PD-1 protein is a coinhibitory receptor that belongs to the cluster of differentiation 28 (CD28) family expressed on T cells. The 2 ligands of PD-1 are programmed death-ligand 1 (PD-L1) (B7 homolog 1 [B7-H1], CD274) and PD-L2 (B7-DC, CD273).
Fig. 1
PD-1 pathway signaling and the targeting of the PD-1 pathway during cancer immunotherapy. T cells recognize the MHC-antigen complex through TCR. CD28 binds to CD80/86 and mediates an activation signal through the PI3K or Ras pathways. PD-1 is ligated with PD-L1 recruitments of SHP-2, which dephosphorylates the downstream molecules and blocks T cell activation. The PD-1 and PD-L1 blockade with mAbs can enhance T cell antitumor activity [28].
APC, antigen-presenting cell; CD, cluster of differentiation; mAbs, monoclonal antibodies; MHC, major histocompatibility complex; PD-1, programmed death-1; PD-L1, programmed death-ligand 1; PI3K, phosphatidylinositol 3-kinase; SHP-2, Src homology region 2 domain-containing phosphatase-2; TCR, T cell receptor.
PD-L1 is a 290-aa type-I transmembrane protein that consists of an immunoglobulin variable (IgV)-like domain, immunoglobulin constant (IgC)-like domain, signal sequence, the transmembrane domain, and intracellular domain [8]. PD-L1 is often expressed by activated cells including T cells, B cells, NK cells, DCs, monocytes/macrophages, activated vascular endothelial cells, mesenchymal stem cells, and cultured bone-marrow-derived mast cells (BMMCs) [30]. PD-L1 is also expressed in human carcinomas of the lung, ovary, colon, and melanomas [31].
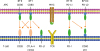
PD-L2 is also a type-I transmembrane protein and is composed of a signal sequence, IgV-like domain, IgC-like domain, stalk, the transmembrane domain, and the cytoplasmic domain. The PD-L2 expression is much more restricted than that of PD-L1; it is only inducibly expressed on macrophages, DCs, and BMMCs [8] and is expressed on 50%–70% of resting peritoneal B1 cells [32]. Although the interaction between PD-1 and PD-L2 shows a 2-6-fold higher affinity compared with the interaction between PD-1 and PD-L1, however, PD-L1 is regarded the primary ligand of PD-1 [33]. The PD-1 pathway-related molecules are displayed in Fig. 2.
Fig. 2
Primary immune checkpoint inhibitors in cells. T cells recognize the MHC-antigen complex through TCR. The ligation of PD-1 with PD-L1/PD-L2 triggers a negative signal in T cells. CTLA-4 competes with CD28 to bind to the costimulatory molecules B7-1 and B7-2 on APCs, mediating an inhibitory signal that leads to the suppression of T cell activation. IgV-like regions are depicted in blue and IgC-like regions in green.
APC, antigen-presenting cell; CD, cluster of differentiation; CTLA-4, cytotoxic T-lymphocyte-associated antigen-4; IgC, immunoglobulin constant; IgV, immunoglobulin variable; MHC, major histocompatibility complex; PD-1, programmed death-1; PD-L1, programmed death-ligand 1; PD-L2, programmed death-ligand 2; TCR, T cell receptor.
LOCAL IMMUNITY OF OC
The local immunity of a tumor indicates that immune cells are present and fulfilling their roles around or within the tumor site rather than being present in the systemic blood circulation [34]. The local infiltration of tumor-antigen-specific T cells in the OC microenvironment was first reported in 1991 [35]. Since then, numerous studies have demonstrated the relationship between tumor-infiltrating lymphocytes (TILs) and the survival of OC patients, and it is widely believed that TILs significantly predict longer survival in OC patients. For example, Zhang et al. [36] reported that patients with intraepithelial T cells had longer PFS and OS than patients without intraepithelial T cells. Although both groups initially exhibited a complete response (CR) to chemotherapy, the 5-year survival rates of the 2 groups (38.0% vs. 4.5%, respectively) were dramatically different. Independent studies on ethnically and geographically diverse populations have also implied that intratumoral CD8+ or CD3+ T cells are significantly associated with patients' OS and PFS, and TIL has been shown to be an active prognostic biomarker of OC [373839]. This understanding of OC immunogenicity has led to the development of immunotherapeutic strategies for OC.
In addition to cancer immunosurveillance, tumor cells can become immune-resistant cells and thus escape human immunosurveillance, a process called immune evasion. Some studies have demonstrated immune evasion mechanisms in patients with OC [4041]. These mechanisms include the loss of tumor antigen expression; down-regulation of major histocompatibility complex (MHC); overexpression of the antiapoptotic effector B-cell lymphoma 2 (BCL-2); increase in the number of immunosuppressive regulatory cells (Tregs, MDSCs); and expression of inhibitory cell surface molecules such as CTLA-4, PD-L1, and Fas ligand, which can kill cytotoxic CD8+ T cells directly [42].
The aforementioned theories and studies suggest that in OC, TILs are associated with increased survival, whereas immune evasion mechanisms are associated with decreased survival. Therefore, immunotherapeutic strategies aimed at improving a host's immune response or reducing immunosuppression are crucial to suppressing OC.
IMMUNE CHECKPOINT INHIBITOR PD-1/PD-L1 IN OC
The current immunotherapies for malignant tumors include therapeutic vaccines, adoptive T cell transfer, immune modulators, cytokines, and immune checkpoint inhibitors [43]. One of the most promising treatments for solid tumors seems to be the blockade of immune checkpoints. Immune checkpoints are inhibitory signals that down-regulate T cell activity to prevent collateral self-tissue damage. However, during tumorigenesis, malignant tumor cells generate abnormal proteins such as cyclooxygenase 2, transforming growth factor-beta, interleukin 10 (IL-10), prostaglandin E2, and xeroderma pigmentosum group A protein that activate immune checkpoint signals and cause immunosuppression. Increased abnormal proteins can suppress the function of intratumoral cytotoxic CD8+ T cells, thereby contributing to tumor immune escape [44454647]. In the immune checkpoint pathway, PD-1-PD-L1/PD-L2 ligation induces the phosphorylation of intracellular tyrosines in ITIMs and ITSMs, which down-regulates B-cell lymphoma-extra large (BCL-XL) expression and regulates T cell differentiation to deliver inhibitory signals into T cells [8]. Blockade of the PD-1 pathway can reverse this inhibitory signal.
In immunogenic OC [353648], the blocking of PD-1/PD-L1 using various agents has been investigated, as shown in Table 1. In September 2014, pembrolizumab (anti-PD-1 antibody, Keytruda®; Merck & Co., Inc., Kenilworth, NJ, USA) became the first agent to be approved by the FDA for the treatment of unresectable stage III/IV metastatic melanoma. In one multicohort, nonrandomized, phase-Ib study (NCT02054806), 26 OC patients were treated with pembrolizumab [49]. The objective response rate of the patients was 11.5%, with 1 CR, 2 partial responses, and 6 stable disease cases. Of the 3 patients who responded, all remained in response after ≥24 weeks at the time of analysis. Nivolumab is another anti-PD-1 antibody and it was approved by the FDA in December 2014. In one study that used nivolumab to treat platinum-resistant OC, the overall response was 15%, including 2 patients with a durable CR; moreover, the disease control rate in all 20 patients was 45%. At the study's termination, the median PFS was 3.5 months and the median OS was 20.0 months. In summary, the first study evaluating nivolumab for the treatment of platinum-resistant OC demonstrated encouraging safety and clinical efficacy [23].
Table 1
PD-1/PD-L1 inhibitors in OC

Studies have shown that the expression level of PD-1/PD-L1 differs across various histological types of OC. Webb et al. [50] found that the percentage of PD-L1-positive tumor cells was lower for the mucinous (26.7%), endometrioid (24.0%), and clear cell (16.2%) subtypes than for the serous subtype (54.4%). Among serous OC cases, the percentage of PD-L1-positive tumor cells increased with the grade (p=0.011). This tendency provides some clues as to which potential OC population will benefit the most from treatment. Moreover, it was discovered that the expression level of PD-1/PD-L1 is related to the prognosis of OC. Hamanishi et al. [37] first reported that a higher expression level of PD-L1 on OC cells predicts a significantly poorer prognosis than a lower expression level (p=0.016). Another study confirmed that PD-L1 expression is associated with poorer prognosis in OC patients and that PD-L1 expression promotes the peritoneal dissemination of OC through host immunosuppression [51].
COMBINED IMMUNOTHERAPIES FOR OC
Silencing the function of PD-1 by using its antibody can reverse immunosuppression and might serve as a promising clinical strategy for controlling malignancies [3137]. However, malignant cells usually acquire tolerance in order to evade host immunity. Furthermore, advanced cancer cannot initiate an optimal immune response, or the magnitude of its response is limited by the extent of the disease burden [18]. In addition, a PD-1/PD-L1 blockade can suppress some tumors, but poorly immunogenic tumors such as ID8 OC do not respond to antibody therapy alone. One study discovered that none of the 3 mAbs (PD-1, CTLA-4, or CD137) prolonged mice survival when administered alone; however, administering a combination of the 3 mAbs (CD137+PD-1+CTLA-4) prolonged the survival of mice with ID8 OC (mean±standard error) from 24.2±1.7 days to 74.4±24.4 days, and 1 of the mice was found to be tumor-free when euthanized 140 days after the first treatment [17]. Based on these findings, the researchers proposed that advanced cancers might respond to combined immunotherapies. An increasing number of studies have demonstrated that the combination of mAbs with chemotherapy, radiotherapy, targeted molecules, or other methods can modify and improve immune response in vivo [192021222324]. These combined immunotherapies have synergistic effects and may radically change tumor treatment paradigms. The clinical combination therapies actively used for OC are listed in Table 2.
Table 2
Combination therapies using checkpoint inhibitors for OC*

aPD, anti-programmed death-1; aCD, anti-cluster of differentiation; aCTLA-4, anti-cytotoxic T-lymphocyte-associated antigen-4; BTKi, Bruton's tyrosine kinase inhibitor; CSF1Ri, colony-stimulating factor 1 receptor inhibitor; CTLA-4, cytotoxic T-lymphocyte-associated antigen-4; DACi, deacetylase inhibitor; EORTC, European Organisation for Research and Treatment of Cancer; FGFR, fibroblast growth factor receptor; mAb, monoclonal antibody; NCI, National Cancer Institute; NCT, National Clinical Trial; OC, ovarian cancer; pan-RAFi, pan-RAF inhibitor; PARPi, poly (adenosine diphosphate [ADP]-ribose) polymerase inhibitor; PD-1, programmed death-1; PD-L1, programmed death-ligand 1; PDGFRi, platelet-derived growth factor receptor inhibitor; PLD, pegylated liposomal doxorubicin; RETi, rearranged during transfection inhibitor; TIL, tumor-infiltrating lymphocyte; VEGFi, vascular endothelial growth factor inhibitor; VEGFR, vascular endothelial growth factor receptor; VISTA, V-domain immunoglobulin suppressor of T cell activation.
*ClinicalTrials.gov. https://clinicaltrials.gov.
1. PD-1 pathway blockade with chemotherapy
Chemoimmunotherapies are attractive, not only because they directly lead to the apoptosis of tumor cells and induce the release of antigens that can drive immune responses, but also because they suppress essential immune regulatory mechanisms that prevent the development of immunity [52]. One previous study revealed that chemotherapeutic agents including paclitaxel, etoposide, and 5-fluorouracil can induce or increase PD-L1 expression on human breast cancer cell lines [53]. Both paclitaxel and etoposide induced dose-dependent increases in PD-L1 surface expression in the human breast cancer cell line MDA-MB-468, whereas 5-fluorouracil was able to induce PD-L1 surface expression in breast cancer cell lines MDA-MB-468 and MDA-MB-435. Induced or increased PD-L1 expression has been considered the prerequisite for the clinical effectiveness of anti-PD-1 immunotherapy.
Currently, platinum-taxane-based adjuvant chemotherapy is the first-line treatment for OC after surgery. Cisplatin promotes its cytotoxicity by forming DNA-protein cross-links, DNA monoadducts, and intrastrand DNA cross-links, all of which trigger cell apoptosis [54]. Cell death can be classified according to the morphological change induced by the lethal process (apoptotic, necrotic, autophagic, or related to mitosis), enzymological criteria (the involvement of nucleases or different types of proteases), functional aspects (programmed or accidental and physiological or pathological), and immunological characteristics (immunogenic or nonimmunogenic) [55]. Although cisplatin does not induce immunogenic tumor cell death, Qin et al. [56] demonstrated that cisplatin can upregulate PD-L1 expression on hepatoma H22 cells; thus, PD-1/PD-L1 expression may represent a promising target for immunotherapy. Unlike cisplatin, oxaliplatin can lead to immunogenic tumor cell death, which is characterized by the exposure of chaperones—including calreticulin, shock proteins, or both—on the cell surface. The cell surface molecules then determine the uptake of tumor antigens and affect the maturation of DCs [55]. Paclitaxel, a member of the taxane family, not only selectively arrests the cell cycle in the M phase, but also inhibits the function of the apoptosis inhibitor protein BCL-2 [57]. In one study of stage II/III breast cancer, increased T cell blastogenesis and NK cell cytotoxicity was found in patients who received taxane treatment compared with patients who did not receive taxane treatment [58]. Moreover, in an OC mouse model, paclitaxel increased the number of CD8+ T cells in the tumor site, upregulated PD-L1 expression, and activated nuclear factor-κB signaling. Tumor-bearing mice treated with a combination of paclitaxel and PD-L1/PD-1 signal blockade were discovered to survive longer than mice treated with paclitaxel alone [20].
2. PD-1 pathway blockade with target therapy
BRCA1 and BRCA2 genes are important components of the homologous recombination pathway. Approximately 17% and 6% of patients with high-grade serous carcinoma (HGSC) have been estimated to exhibit germline and somatic mutations in these genes, respectively [59]. Poly (adenosine diphosphate [ADP]-ribose) polymerase (PARP) plays a significant role in single-stranded DNA break repair and genomic stability through the base excision repair pathway [60]. PARP inhibition causes the death of BRCA-mutated cells through the synthetic lethality mechanism. Olaparib (Lynparza™; AstraZeneca Pharmaceuticals LP, Wilmington, DE, USA) is a PARP inhibitor that was approved by the FDA in December 2014 for the treatment of patients with germline mutations in BRCA1/2 [61]. One previous study showed that BRCA1- and BRCA2-mutated OC tumors exhibit increased CD3+ and CD8+ TILs and increased PD-1/PD-L1 expression on tumor-associated immune cells [21]. Mechanistically, PARP inhibitors inactivate glycogen synthase kinase-3β, which in turn improves PARP-inhibitor-mediated PD-L1 upregulation. Accordingly, PD-L1 pathway blockade resensitizes PARP-inhibitor-treated tumor cells to T cell killing. The combined use of a PARP inhibitor and anti-PD-L1 immunotherapy can significantly increase therapeutic efficacy for breast cancer compared with each agent alone [62]. Therefore, BRCA1- and BRCA2-mutated OC patients may be a suitable candidate population for these inhibitors.
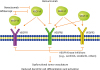
Bevacizumab is a humanized mAb that inhibits angiogenesis by blocking the binding of vascular endothelial growth factor (VEGF)-A to VEGF receptor 1 (VEGFR1) and VEGFR2. It has been demonstrated that VEGF/VEGFR signaling can result in dysfunctional vasculature that inhibits the infiltration of T cells into the tumor site and reduces DC differentiation and activation, which may impair T cell priming. In addition to its activity on tumor vasculature for modulating host immunity, VEGF-A exerts direct inhibitory effects on immune cells, such as increases in the expression of checkpoint molecules on CD8+ T cells and the modulation of proliferation of Tregs. Treatment of mouse models of cancers such as colorectal cancer, lung cancer, cervical cancer, and OC with inhibitors of VEGF-A/VEGFR signaling increases T cell recruitment and infiltration into the tumor site, and it may be synergistic with anti-PD-1 therapy [63]. One study found that simultaneous treatment with anti-PD-1 and anti-VEGFR2 mAbs inhibited tumor growth synergistically in a mice model of colon-26 adenocarcinoma without overt toxicity [64]. An ongoing phase-II study is evaluating the safety and efficacy of PD-L1 in combination with bevacizumab in recurrent platinum-resistant OC. VEGF/VEGFR signaling pathways and the approved therapeutic agents targeting these pathways are illustrated in Fig. 3.
Fig. 3
VEGF/VEGFR signaling and the approved therapeutic agents targeting this signaling. VEGF/VEGFR signaling can lead to dysfunctional vasculature that inhibits the infiltration of T cells into the tumor site and reduces DC differentiation and activation. Inhibition of VEGF/VEGFR signaling can improve intratumoral immune cell infiltration and antitumor immune responses [63].
DC, dendritic cell; VEGF, vascular endothelial growth factor; VEGFR, vascular endothelial growth factor receptor.
Epidermal growth factor receptor (EGFR) is a tyrosine kinase receptor, and the binding of EGF to EGFR induces the tyrosine phosphorylation of diverse cellular proteins. Increased EGFR expression in tumors indicates that the tumors are more aggressive, more likely to metastasize, and more resistant to chemotherapy, which lead to poorer outcomes in cancer patients. Cetuximab (Erbitux®; Merck & Co., Inc.) binds to the extracellular domain of EGFR, inhibiting EGFR signaling and accelerating receptor internalization [65]. Treatment of OC cell lines in vitro with cetuximab suppresses cell growth, potentiates cell apoptosis, and impairs tumor metastasis [66]. Multiple trials are investigating strategies for the combined application of cetuximab with anti-PD-1/PD-L1 agents in various cancers, such as recurrent or metastatic squamous cell cancer of the head and neck (NCT02358031) and metastatic colorectal cancer (NCT02934529).
According to the dualistic model of carcinogenesis, epithelial OC can be categorized as type I and type II. Type I cancers consist of low-grade serous, low-grade endometrioid, clear cell, and mucinous carcinomas, as well as Brenner tumors. They are relatively indolent, present in early stages, and include specific mutations (KRAS, BRAF, ERBB2, CTNNB1, PTEN, PIK3CA, ARID1A, and PPPR1A) targeting specific cell signaling pathways. Type II cancers consist of HGSC, high-grade endometrioid, malignant mixed mesodermal tumors, and undifferentiated carcinomas. They are relatively aggressive, present in advanced stages, and harbor a high frequency of TP53 mutations [67]. Next generation sequencing (NGS) technology can be used for whole-exome and whole-genome sequencing. Studies have demonstrated that patients with a high frequency of somatic mutations are more likely to benefit from treatment with PD-1 inhibitors. The enhanced mutation load may activate adaptive immunity and attract CD8+ cell infiltrates. Thus, genomic analysis of the total mutational load using NGS can be employed to determine the population that will benefit from combined immunotherapy [68].
3. PD-1 pathway blockade with radiotherapy
Ionizing irradiation is one of the most common treatment strategies for cancer. Radiation predominantly induces DNA damage in tumor cells through base damage, base release, depolymerization, crosslinking, and strand breakage, consequently leading to the apoptosis, necrosis, mitotic catastrophe, autophagy, or senescence of the cells [2269]. Following radiotherapy, cancer cells release various substances such as IL-6, IL-8, and tumor necrosis factor (TNF)-α, which can stimulate the immune system [22]. Zeng et al. [70] found that anti-PD-1 immunotherapy combined with stereotactic radiotherapy significantly prolonged the survival of glioma-tumor-bearing mice and generated long-term antitumor memory. Testing of long-term antitumor memory revealed that when naïve and “cured” mice (animals surviving >90 days after intracranial tumor implantation in combined immunotherapy group) were rechallenged using flank injections of GL261-luc cells, none of the “cured” mice had developed tumors by day 60 after implantation whereas 100% (8/8) of the naïve mice had developed flank tumors of size >1,000 mm3 by day 20 after implantation. The release of diverse tumor-associated antigens in a proinflammatory environment has been speculated to act as a vaccine, leading to the generation of immunologic memory. In melanoma, colorectal, or breast cancer cell lines, low doses of fractionated radiotherapy were demonstrated to lead to PD-L1 upregulation on tumor cells. Notably, fractionated radiotherapy combined with PD-1 or PD-L1 mAbs produced efficacious CD8+ T cell immune responses that improved long-term survival and protected against tumor rechallenge [71]. In OC cell lines, high doses of gamma irradiation (5,000–10,000 cGy) were confirmed to induce a significant and long-lasting upregulation of MHC class I (MHC I), MHC II, and antigens (CA125 and Her2-neu) expressed on the OC cell lines. The enhancement of antigen expression, which was crucial for both the recognition and destruction of OC cells by the host immune system, was persistent until all cells had died [72]. Deng et al. [73] reported that radiotherapy combined with anti-PD-L1 immunotherapy reduced the number of MDSCs, which is characterized by the surface makers of CD11b+ and Gr-1+, thus reducing the suppressive effects on the immune system. Therefore, the combination of immunotherapy with radiotherapy and PD-1 signaling blockade may be an effective antitumor strategy for improving treatment outcomes for cancers including OC.
4. PD-1 pathway blockade with anti-CTLA-4 mAb
CTLA-4 (also known as CD152) was identified in 1987 as the first coinhibitory molecule that plays a significant role in the down-regulation of T cell activity in vivo to limit self-damage [74]. CTLA-4 has a high affinity for CD86/CD80 and can thus effectively outcompete CD28 (a costimulatory molecule) for its ligands and deliver inhibitory signals to T cells [75] (Fig. 2). The precise mechanism of CTLA-4-mediated suppressive signaling is currently being explored. CTLA-4 has been postulated to suppress T cell function by inducing the dephosphorylation of the ζ chain of the TCR complex, through the Lck-dependent recruitment of SHP-2 [76]. Another study reported that CTLA-4 also suppresses T cell function by inhibiting signals from CD3-ε and in vitro evidence suggested that TCR-ζ and CD3-ε chains exert different effects as a result of heterogeneity in the primary sequence of their immune receptor tyrosine-based motifs [77]. TCR chains are physically associated with a set of invariant molecules: CD3-γ, CD3-δ, CD3-ε, and the TCR-ζ and η chains. The TCR-ζ chain plays a vital role in linking TCR-triggering to several protein tyrosine kinases—such as ZAP-70, Syk, Lck, and Fyn—which in turn control signal transduction pathways that regulate lymphokine and lymphokine receptor gene expression [78]. CD3-ε is critical in T cell thymic differentiation and functional competence [7980]. The administration of therapeutic mAbs to block CTLA-4 can prevent immune inactivation through the reversion of the aforementioned mechanisms and induce rational antitumor immune responses through the inhibition of Treg cell function and enhancement of effector CD4+ T cell activity [81]. Moreover, CTLA-4 and the PD-1 pathway are believed to operate during different stages of an immune response. CTLA-4 regulates T cell proliferation early and mainly in lymph nodes, whereas the PD-1 pathway suppresses T cell proliferation later and primarily in peripheral tissues [82]. These key similarities (both can induce immunosuppression) and differences (stages of immune response) of CTLA-4 and the PD-1 pathway indicate the possibility of their combination for cancer therapy. To date, 2 CTLA-4-targeting agents (tremelimumab and ipilimumab) have been assessed in clinical trials [83].
Tremelimumab is a fully humanized immunoglobulin G2 (IgG2) mAb that stimulates the proliferation of CD4+ T cells, IL-2 production by CD4+ T cells, and the proliferation of CD8+ cells [8485]. Tremelimumab blocks the binding of CD80/CD86 to CTLA-4, inhibits CTLA-4-mediated down-regulation of T cell activation, and enhances anticancer immunity through the potential aforementioned mechanism. Ipilimumab is a fully humanized IgG1-kappa recombinant mAb that inhibits CTLA-4; it was approved by the FDA in 2011 for clinical use in metastatic melanoma patients [83].
PD-1 and CTLA-4 are inhibitory cell surface molecules, and their blockade has been found to have promising efficiency in various solid tumors such as colon carcinoma and OC. Blockade of both PD-1 and CTLA-4 was previously shown to lead to tumor rejection in 2/3 of mice [85]. The double blockade was associated with the increased proliferation of antigen-specific effector CD8+ and CD4+ T cells, antigen-specific cytokine release (granzyme B, interferon (IFN)-γ, TNF-α), and the inhibition of the suppressive functions of Tregs. A previous study demonstrated that in the presence of anti-CTLA-4 mAb, spleen cells exhibited improved production of IL-2 and IFN-γ in mice with early fibrosarcoma and OC. Moreover, the mixed lymphocyte reaction (MLR) calculated from the uptake of 3H thymidine showed that the level of T cell proliferation increased with the dose of anti-CTLA-4 mAb. The administration of anti-CTLA-4 mAb at a high dose (20 µg/mL) markedly enhanced MLR [86]. Because preclinical data has demonstrated the synergistic efficacy of a combined PD-1/CTLA-4 blockade, the focus of research has shifted to the combined immunotherapy of the PD-1/CTLA-4 blockade [87]. Several clinical trials investigating the anti-PD-1/anti-PD-L1 antibody and anti-CTLA-4 antibody have identified marked clinical effects in numerous solid tumors [88]. In patients with melanoma, a total of 53 patients received concurrent therapy with nivolumab (anti-PD-1 mAb) and ipilimumab (anti-CTLA-4 mAb). The objective response rate in the concurrent-regimen group was 40%, with a manageable safety profile. The combined immunotherapy demonstrated more rapid and deeper tumor regression in a substantial proportion of patients than has been reported in published data on monotherapy [88]. Based on preclinical studies on OC and clinical trials of other malignancies treated using combined immunotherapy, there is an ongoing phase-II study using nivolumab both alone and in combination with ipilimumab in recurrent epithelial OC that aims to identify the objective tumor response rate (NCT02498600) [89]. Other ongoing clinical trials are listed in Table 2.
5. PD-1 pathway blockade with other agents
In addition to the aforementioned active agents investigated in clinical trials in OC patients, several new combined applications have been tested in animal models. OX40 (also known as CD134) is a costimulatory molecule belonging to the TNF receptor family and is primarily expressed on activated T cells—including CD4 T, CD8 T, type 1 T helper (Th1), Th2, and Th17 cells—and Tregs. However, naïve CD4, CD8, and most memory T cells do not express OX40 [24]. One study confirmed that the combination of antagonistic anti-PD-1 antibodies with agonistic anti-OX40 antibodies markedly inhibited tumor outgrowth in an OC model by increasing CD4+ and CD8+ T cells and decreasing immunosuppressive CD4+ FoxP3+ Treg cells and CD11b+ Gr-1+ MDSCs [90]. Wei et al. [91] discovered that the combination of anti-PD-1 mAbs and anti-CD137 mAbs doubled the OS. Mice with OC that were treated using this combination exhibited a significantly increased total number of CD8+ T cells, both in peritoneal lavage and spleens, which were determined to be functional using their antigen-specific cytolytic activity and IFN-γ production. Moreover, adding cisplatin to the combination of the 2 mAbs increased OS by more than 90 days. Lu et al. [92] discovered that the costimulatory receptor glucocorticoid-induced TNFR-related protein (GITR) is upregulated on activated CD4+ and CD8+ T cells; GITR engagement increases the proliferation and activation of these T cells and the production of cytokines such as IFN-γ by these T cells. Concomitant PD-1 blockade and GITR triggering can remarkably inhibit ID8 ovarian tumor growth and synergistically improve OS in mice. The durable antitumor effect was associated with the increased frequency of IFN-γ-producing effector T cells and decreased immunosuppressive Tregs and MDSCs, which changed an immunosuppressive milieu to an immunostimulatory state in the peritoneal cavity. In addition, combined treatment induced an antigen-specific immune response, as evidenced by antigen-specific IFN-γ production and the cytolytic activity of spleen cells in treated mice. Moreover, treatment with anti-PD-1/GITR mAb combined with chemotherapeutic drugs (cisplatin or paclitaxel) further increases the antitumor efficacy. Although the detailed mechanisms of synergy between combined immunotherapy and cisplatin/paclitaxel remain unclear, it is speculated that their synergy is partially due to increased tumor antigenicity and the chemotherapy-induced death of immunosuppressive cells. Trabectedin (ET-743, Yondelis®; Janssen Products, LP, Beerse, Belgium) is a synthetic, marine-derived anticancer agent that can arrest the cell cycle and is currently used to treat patients with relapsed OC. Trabectedin binds to the minor groove of duplex DNA, bends the DNA helix to the major groove, and causes perturbation of the cell cycle [93]. Tumor cells in the G1 phase are more susceptible to being delayed by trabectedin than tumor cells in the S and G2+M phases [9495]. Treatment of mice with OC using combined α-PD-1 mAb and trabectedin induced a strong antitumor immune response, resulting in notable tumor inhibition; half of these mice were tumor-free for 90 days after OC cell inoculation. A mechanistic investigation revealed that the combination treatment remarkably increased the percentage of effector CD4+ and CD8+ T cells and concomitantly decreased that of immunosuppressive Tregs and MDSCs, resulting in a substantial elevation of the ratios of effector CD4+ and CD8+ T cells to immunosuppressive Tregs and MDSCs. Moreover, the combined α-PD-1 mAb and trabectedin treatment was also found to alter the immune microenvironment by increasing Th1 effector T cell recruitment and functionality (i.e., increased transcripts for IFN-γ, IL-12p40, and T-bet) [96].
THE ADVERSE IMMUNOLOGICAL EFFECTS OF COMBINED IMMUNOTHERAPY
The data obtained to date have indicated that combining anti-PD-1 mAbs with chemotherapy, radiotherapy, target therapy, or immunotherapy results in not only increased antitumor effects in OC (e.g., an increased number of effector CD8+ T cells in the tumor site, increased IFN-γ level, and prolonged OS) [2086], but also markedly increased toxicity. In particular, combined immunotherapies are associated with a unique set of autoimmune side effects, termed immune-related adverse events (irAEs). These adverse events are different from the toxicity observed in conventional antitumor chemotherapy. Because anti-PD-1 mAbs nonspecifically activate T cells, immune-mediated damage of tissue or irAEs can occur. In clinical trials, the most common irAEs have included hypophysitis, colitis, hepatitis, pneumonitis, and rash. Some patients may also experience irAEs resembling inflammatory and rheumatic diseases such as arthritis, nephritis, myositis, and polymyalgia-like syndromes [97]. Biopsies of patients with colitis have discovered various inflammatory cell infiltrates; moreover, the development of colitis is associated with an elevated level of IL-17, highlighting the relationship between immune activation and tissue damage [98]. IrAEs are serious toxicities about which the FDA has posted precautions and warnings [99100].
IrAEs are usually reversible but may occasionally be severe or fatal, and the timing of their onset varies. They can occur in any organ or tissue. The clinical presentations of irAEs in the gastrointestinal system include colitis, diarrhea, abdominal pain, blood or mucus in the stool, and bowel perforation, and these irAEs can be treated with antidiarrheals, oral hydration, electrolyte substitution, and budesonide. The irAEs occurring in the skin include rash, pruritus, psoriasis, and leukocytoclastic vasculitis, which are treated using topical steroids, urea-containing creams, and antihistamines. Other systems, including the nervous, endocrine, urinary, ophthalmic, and respiratory systems, may also experience irAEs, which are alleviated by supportive care and corticosteroids [99]. It is of great importance for patients that caregivers and clinical teams should detect the early signs and symptoms of irAEs and manage them successfully according to their clinical features. The timely recognition of irAEs and their intervention using immunosuppression based on the severity of the events usually enables complete reversibility; conversely, the failure to provide timely intervention may result in severe toxicity or even death.
One meta-analysis demonstrated that, among patients with melanoma, the PD-1 inhibitor group showed a significantly lower rate of grade 3–4 adverse effects than the chemotherapy group (p<0.001) [101]. Another meta-analysis revealed that, although immunotherapies may induce various irAEs, PD-1/PD-L1 blockade generally induces objective responses with a tolerable adverse effect profile [102].
CONCLUSION
Recent years have witnessed the rapid development and successful application of combined immunotherapy in the fight against OC. However, several emerging issues will need to be addressed in clinical practice, such as the dosage and sequence for optimal synergy, the different immunotherapeutic response in different subtypes of OC, and the infertility risk in young women exposed to PD-1/PD-L1 blockade. Currently, the mechanisms of combined immunotherapy for OC remain to be defined, but the outcomes seem promising. We suggest that an increasing number of successive clinical trials on combined immunotherapy, along with a comprehensive investigation of the mechanism of cancer immunity, will enable the wider clinical application of combined immunotherapy in the near future.
 XML Download
XML Download