

 PDF
PDF Citation
Citation Print
Print



INTRODUCTION
Clear cell carcinoma (CCC) of the ovary, which was first recognized by the World Health Organization as a distinct histologic subtype in 1973 [1]. There are marked geographic differences in the prevalence of ovarian CCC. According to recent Surveillance, Epidemiology, and End Results (SEER) data revealed that the incidence of ovarian CCC in women living in United States is 4.8% in whites, 3.1% in blacks, and 11.1% in Asians [2]. In Japan, the prevalence of ovarian CCC is higher than in western countries, with an estimated incidence of 25% of epithelial ovarian cancers [3].
CCC of the ovary is known to be less sensitive to platinum-based front-line chemotherapy and to be associated with a worse prognosis than serous adenocarcinoma (SAC) or endometrioid adenocarcinoma [4567]. On the basis of previous preclinical and clinical studies suggesting that irinotecan is more effective against ovarian CCC than any other anticancer agents [89], a phase III study comparing the activity of irinotecan plus cisplatin versus carboplatin plus paclitaxel as a front-line treatment for ovarian CCC was conducted by the Japanese Gynecologic Oncology Group (JGOG; protocol JGOG3017). However, this study failed to demonstrate the superiority of irinotecan plus cisplatin over carboplatin plus paclitaxel [10].
Another important problem in the clinical management of ovarian CCC is the lack of effective chemotherapy for recurrent disease after front-line treatment with platinum-based chemotherapy [1112]. A previous report showed that the response rate for various regimens in the setting of second-line chemotherapy for recurrent platinum-resistant ovarian CCC was only 1% [11]. Moreover, previous investigations suggested that platinum sensitivity defined using platinum-free interval does not predict the treatment outcomes in recurrent ovarian CCC [12, 13]. Therefore, to improve the survival of patients with ovarian CCC, the deeper understanding of the mechanism of carcinogenesis and chemoresistance as well as the development of novel treatment strategies in the setting of both front-line treatment and salvage treatment for recurrent disease are needed.
In this article, by understanding the mechanism responsible for the carcinogenesis, we first summarize the promising therapeutic targets in ovarian CCC. Then, we discuss how these therapeutic targets might best be targeted as treatments for ovarian CCC in light of ongoing preclinical and clinical trials.
MECHANISM OF CCC CARCINOGENESIS
1. Genetic changes in ovarian CCC
Unlike high-grade SAC, ovarian CCC are generally p53 wild type and have a lower frequency of breast cancer 1 (BRCA1) and BRCA2 mutations [1415]. Table 1 summarizes the critical genetic alterations observed in ovarian CCC [151617181920212223]. The most frequent and important alterations are AT rich interactive domain 1A (ARID1A) and phosphatidylinositol-45-bisphosphate 3-kinase catalytic subunit α (PIK3CA) mutations [1617]. It has been reported that tumors with ARID1A mutations also frequently harbor phosphatase and tensin homolog (PTEN) or PIK3CA mutations, suggesting their collaboration in CCC tumorigenesis [24]. Consistent with this, recent investigations involving genetically engineered mouse model (GEM) demonstrated that inactivation of ARID1A alone is insufficient for tumor initiation; it requires additional genetic alterations such as PIK3CA to drive CCC tumorigenesis [25].
Table 1
Critical genetic changes in clear cell carcinoma

| Gene | Gene type | Change | Pathways affected | Roles in tumor development | Reference |
|---|---|---|---|---|---|
| ARID1A | Tumor suppressor | Mutation in ~50% | SWI/SNF chromatin remodeling complex | Modulate accessibility of transcription factors to promoters | [16] |
| PIK3CA | Oncogenic | Mutation in ~40% | PI3K/AKT/mTOR | Proliferation/survival | [17] |
| PPP2R1A | Oncogenic | Mutation in 7% | AKT/MAPK | Proliferation/survival | [18] |
| KRAS | Oncogenic | Mutation in 5% | AKT/MAPK | Proliferation/survival | [19] |
| BRCA1/2 | Tumor suppressor | Mutation in 6% | DNA repair | Genomic instability | [15] |
| PTEN | Tumor suppressor | Mutation in 5% | PI3K/AKT/mTOR | Proliferation/survival | [20] |
| CDKN2A/2B | Tumor suppressor | Deletion in 9% | CDK inhibitors (p15/p16) | Cell cycle progression | [21] |
| ZNF217 | Oncogenic | Amplification in 36% | ZNF217 | Antiapoptosis | [21] |
| PPM1D | Oncogenic | Amplification in 10% | p53 mediated apoptosis | Antiapoptosis | [22] |
| AKT2 | Oncogenic | Amplification in 14% | AKT/mTOR | Proliferation/survival | [23] |
| MET | Oncogene | Amplification in 37% | AKT/MAPK | Proliferation/survival | [23] |
ARID1A, AT rich interactive domain 1A; BRCA, breast cancer; CDK, cyclin-dependent kinase; CDKN, cyclin-dependent kinase inhibitor; MAPK, mitogen-activated protein kinases; mTOR, mammalian target of rapamycin; PIK3CA, phosphatidylinositol-45-bisphosphate 3-kinase catalytic subunit alpha; PI3K, phosphatidylinositol 3-kinase; PPM1D, protein phosphatase 1D; PPP2R1A, protein phosphatase 2 regulatory subunits 1A; PTEN, phosphatase and tensin homolog; SWI/SNF, SWItch/Sucrose Non-Fermentable; ZNF217, zinc finger protein 217.
2. Altered protein expressions
The protein expressions in ovarian CCC have been intensively investigated [232627282930313233343536]. Table 2 is listing the proteins highly expressed in ovarian CCC. Interestingly, these proteins can be classified into five pathways: phosphatidylinositol 3-kinase (PI3K)/AKT/mammalian target of rapamycin (mTOR) pathway, hypoxia-inducible factor 1α (HIF-1α)/vascular endothelial growth factor (VEGF) pathway, hepatocyte nuclear factor 1β (HNF-1β) pathway, interleukin 6 (IL-6)/signal transducer and activator of transcription 3 (STAT3) pathway and MET pathway. These pathways may be important targets for the treatment of ovarian CCC, and inhibitors targeting some of these pathways are in clinical development.
Table 2
Proteins highly expressed or activated in clear cell carcinoma

| Protein | Roles in tumor development | Affected pathways | Reference |
|---|---|---|---|
| AKT | Proliferation/survival | PI3K/AKT/mTOR pathway | [26] |
| mTORC1 | Proliferation/survival/angiogenesis | PI3K/AKT/mTOR pathway | [27] |
| mTORC2 | Proliferation/survival | PI3K/AKT/mTOR pathway | [28] |
| MET | Proliferation/survival | PI3K/AKT/mTOR and Raf/Ras/MAPK pathways | [23] |
| HIF-1β | Angiogenesis | VEGF pathway | [29] |
| VEGF | Angiogenesis | VEGF pathway | [30] |
| HNF-1β | Stimulation of transcription | HNF-1β pathway | [31] |
| Annexin A4 | Detoxification, chemoresistance | HNF-1β pathway | [32] |
| Osteopontin | Survival/migration/invasion | HNF-1β pathway | [33] |
| IGFBP-1 | Proliferation/survival | HNF-1β pathway | [34] |
| IL-6 | Proliferation/angiogenesis | IL-6/STAT3 pathway | [3536] |
| STAT-3 | Proliferation/angiogenesis | IL-6/STAT3 pathway | [3536] |
HIF-1β, hypoxia-inducible factor 1β; HNF-1β, hepatocyte nuclear factor 1β; IGFBP-1, insulin-like growth factor-binding protein 1; IL-6, interleukin 6; MAPK, mitogen-activated protein kinases; mTOR, mammalian target of rapamycin; mTORC1, mammalian target of rapamycin complex 1; mTORC2, mammalian target of rapamycin complex 2; PI3K, phosphatidylinositol 3-kinase; STAT-3, signal transducer and activator of transcription 3; VEGF, vascular endothelial growth factor.
3. Genetically GEM of ovarian CCC
GEM has advantages over nude mice as an experimental model, because the tumors develop spontaneously, the site of tumors is orthotopic, and they are immuno-competent [37]. As shown in Table 3, although p53, PTEN, PIK3CA, and ARID1A are known to be key genes for ovarian carcinogenesis, none of the genes, when mutated individually, could lead to the development of ovarian cancer in mice. However, the combination of at least 2 mutations had resulted in the development of serous, endometrioid, or undifferentiated adenocarcinomas. Very recently, a mouse model of ovarian CCC carrying coexisting ARID1A and PIK3CA mutations has been introduced [25]. By using the intra-bursal gene-delivery method, the authors induced the ARID1A-loss and PIK3CA-hyperactivation, and succeeded in the development of ovarian CCC. Importantly, consistent with the finding from ovarian CCC in human, PI3K/AKT/mTOR pathway is hyperactivated in ovarian CCC in this mouse model. When the mice were treated with a PI3K inhibitor, the survival of mice was significantly prolonged [25]. Additional important finding in this mouse model is the activation of IL-6/STAT3 signaling, which is also consistent with the findings in human CCC [3536]. The precise mechanisms responsible for the IL-6/STAT3 signaling activation remain unknown, but the authors speculated that ARID1A may negatively regulate the expression of IL-6, and oncogenic PIK3CA mutation enhances the IL-6 production in the absence of negative-regulation by ARID1A in ovarian CCC [25]. We believe that the mouse model of ovarian CCC can be used for the response assessment of the candidate drugs, biomarker analyses, mechanistic investigations for the understanding of chemoresistance or carcinogenesis, or chemoprevention study, that can lead to the acceleration of the development of effective therapies against ovarian CCC.
Table 3
Genetically engineered mouse models of ovarian cancer

APC, adenomatosis polyposis coli; Arid1a, AT rich interactive domain 1A; BRCA, breast cancer; CCC, clear cell carcinoma; EMA, endometrioid adenocarcinoma; EOC, Epithelial ovarian cancer; PIK3CA, phosphatidylinositol-45-bisphosphate 3-kinase catalytic subunit alpha; PTEN, phosphatase and tensin homolog; RB, retinoblastoma protein; SAC, serous adenocarcinoma.
PROMISING THERAPEUTIC TARGETS
1. HNF-1β pathway
HNF-1β is a transcriptional factor which is commonly (over 95%) expressed in ovarian CCC [31]. Previous investigations suggested that it regulates the expression of proteins/genes such as annexin A4, uridine diphosphate (UDP)-glucuronosyl transferase 1A1 (UGT1A1), osteopontin, or insulin-like growth factor-binding protein 1 (IGFBP-1) that are important for cancer progression [31]. However, the specific inhibitor of HNF-1β has not been developed yet, and the significance of HNF-1β as a therapeutic target remains to be elucidated. Because of its high expression rate in ovarian CCC [33], HNF-1β is now used as a diagnostic marker of ovarian CCC [38].
2. VEGF and IL-6/STAT3 pathways
Both VEGF and IL-6 bind to their corresponding receptors, activate downstream effectors, and promote the progression of human cancers (Fig. 1).
Fig. 1
(A) Vascular endothelial growth factor (VEGF), (B) interleukin 6 (IL-6)/signal transducer and activator of transcription 3 (STAT3), and (C) MET pathways. HGF, hepatocyte growth factor; MAPK, mitogen-activated protein kinases; MEK, mitogen-activated protein kinase kinase; mTOR, mammalian target of rapamycin; PI3K, phosphatidylinositol 3-kinase.

VEGF promotes proliferation, survival, and migration of endothelial cells to induce tumor angiogenesis, and inhibitors of VEGF or VEGF receptor (VEGFR) have now approved in the treatment of ovarian cancer [39]. It was reported that VEGF is commonly (90% to 100%) expressed in both early-stage and advanced-stage ovarian CCC, and its expression is associated with patient’s survival [30].
IL-6 is a pleiotropic proinflammatory cytokine that has emerged as a mediator of pivotal processes such as cell proliferation, angiogenesis, and chemoresistance within tumor microenvironment. It has been reported that IL-6/STAT3 signaling is frequently activated in ovarian CCC, and IL-6 receptor (IL-6R) expression is associated with survival [3536]. Collectively, these results indicate that the progression of ovarian CCC is dependent, at least in part, on VEGF and IL-6/STAT3 pathways. Currently, the clinical activity of VEGFR-inhibitor sunitinib is being evaluated in a phase II trial in patients with ovarian CCC [40]. Although the final results have not been reported, the data presented in the Society of Gynecologic Oncology 2015 meeting has suggested that sunitinib has minimal clinical activity in patients with recurrent CCC as the second- or third-line treatment, with a response rate of 6.7% [41]. As for IL-6-targeted therapy, no clinical trials have been conducted yet.
3. PI3K/AKT/mTOR pathway
The PI3K/AKT/mTOR pathway plays a critical role in the malignant transformation of human tumors and their subsequent growth, proliferation, and metastasis [42]. Previous studies have shown that ovarian CCC often exhibit genetic alterations in one or more components of the PI3K/AKT/mTOR signaling pathway (Table 1); PIK3CA mutation in 33% to 40% [17], PTEN mutation in 5% [20], or AKT2 amplification in 14% [23]. In addition to these genetic alterations, the loss of PTEN expression has been detected in 40% of ovarian CCC [43]. Consistent with these alterations, our group examined tissue microarrays of 98 primary ovarian tumors (52 CCC and 46 SAC) to tissue microarrays and demonstrated that AKT, mammalian target of rapamycin complex 1 (mTORC1), and mTORC2 are more frequently activated in CCC than SAC (CCC vs. SAC for AKT, 69.2% vs. 63%; mTORC1, 86.6% vs. 50%; and mTORC2, 71.2% vs. 45.7%) [262728]. Importantly, PI3K/AKT/mTOR inhibitors had marked anti-tumor effects in ovarian cancer cells exhibiting high AKT/mTORC1 activity, but minimal effects in ovarian cancer cells displaying low AKT/mTORC1 activity [262728]. Thus, PI3K/AKT/mTOR pathway is regarded as an attractive therapeutic target in in ovarian CCC.
The PI3K/AKT/mTOR pathway inhibitors include PI3K inhibitors, AKT inhibitors, mTORC1 inhibitors, and dual inhibitors (mTORC1/2 inhibitors or PI3K/mTOR inhibitors) (Fig. 2). These inhibitors are currently in various stages of clinical development in the setting of single agent or in combination with other targeted agents in ovarian cancer patients (Table 4). Of these, GOG-0268 is a phase II trial specifically targeting ovarian CCC, and is examining temsirolimus in combination with carboplatin and paclitaxel followed by temsirolimus consolidation as a first-line therapy for patients with stage III to IV diseases. As the dual inhibitors are theoretically more efficacious than the conventional inhibitors (mTORC1 inhibitor or PI3K inhibitors) [28], the clinical activity of dual inhibitors should be investigated in future trials in patients with ovarian CCC.
Fig. 2
Phosphatidylinositol 3-kinase (PI3K)/AKT/mammalian target of rapamycin (mTOR) inhibitors. mTORC1, mammalian target of rapamycin complex 1; mTORC2, mammalian target of rapamycin complex 2.
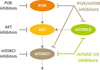
Table 4
Summary of PI3K/AKT inhibitors in clinical trials

mTOR, mammalian target of rapamycin; mTORC1, mammalian target of rapamycin complex 1; mTORC2, mammalian target of rapamycin complex 2; NA, not available; PARP, poly (ADP-ribose) polymerase; PI3K, phosphatidylinositol 3-kinase; VEGF, vascular endothelial growth factor; VEGFR, vascular endothelial growth factor receptor.
*This study is currently being conducted by Yakult Honsha Co. Ltd.
4. MET pathway
MET is a receptor tyrosine kinase that, after binding with hepatocyte growth factor, activates Raf/Ras/mitogen-activated protein kinases (MAPK) and PI3K/AKT/mTOR signaling pathways and stimulates cancer cell proliferation, migration, and invasion (Fig. 1). Recent investigations have suggested that MET overexpression and gene amplification were commonly observed in CCC with their frequencies of 22% and 24% to 37%, respectively [2344]. Moreover, both MET overexpression and gene amplification were shown to be associated with worse prognoses in CCC patients [2545]. Furthermore, MET knockdown in CCC cell lines with MET amplification resulted in both increased apoptosis and senescence in vitro [23]. These results suggest that MET-targeted therapy may have therapeutic efficacy in patients with CCC. Currently, the safety and the activity of MET inhibitor (INC280) are being evaluated in a phase I trial in patients with advanced MET-dependent solid tumor including ovarian cancer [45].
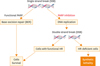
5. Poly (ADP-ribose) polymerase
Poly (ADP-ribose) polymerase (PARP) is a protein that is involved in the repair of single-strand breaks (SSBs) of DNA. When PARP is inhibited, unrepaired SSBs can result in double-strand breaks (DSBs). As DSBs of DNA is repaired mainly by homologous recombination pathway (HR), DSBs induced by PARP-inhibition can lead to lethal DNA damages in HR-deficient cells: i.e., cells with BRCA1/2 mutations (Fig. 3). Consistent with these findings, ovarian cancers occurring in patients with deleterious germline mutations in BRCA1 or BRCA2 have shown significant sensitivity to PARP inhibitors [4647]. It is also becoming clear that germline BRCA1/2 mutations are neither necessary nor sufficient for patients to derive benefit from PARP inhibitors. Several mechanisms that induce HR dysfunction have been identified: i.e., BRCA1 promoter methylation [48], or loss or reduction in proteins necessary for HR. PTEN loss may be a common contributing event causing HR dysfunction [49], and the increased PARP inhibitor susceptibility was demonstrated in a series of cell lines with PTEN mutation [49]. Moreover, previous investigations have demonstrated that PI3K inhibition is associated with HR dysfunction, leading to increased DNA damage and more sensitization of cancer cells to PARP inhibitors [50]. Thus, although the germline mutations in BRCA1 or BRCA2 are observed only in 6% of ovarian CCC patients [15], we consider that the activity of PARP inhibitor plus PI3K/AKT/mTOR inhibitor is worth investigating in this patient population, in which loss of PTEN expression is observed in 40% of cases [43]. Currently, phase I trials of combined treatment involving the PARP inhibitor olaparib and pan-PI3K inhibitors or AKT-inhibitor are enrolling cancer patients including ovarian cancer (Table 4).
KEY CYTOTOXIC AGENTS
1. Nucleotide excision repair system and platinum-resistance
Nucleotide excision repair (NER) is a versatile DNA repair system that recognizes and acts against DNA damage induced by platinum-based agents [51]. According to previous reports, increased NER activity is associated with resistance to platinum-based agents, and NER deficiency is associated with increased sensitivity to platinum-based agents [5253]. Previous study of ovarian cancer showed that NER activity is increased in ovarian CCC than in any other histological subtypes of ovarian cancer [54], indicating that the increased NER activity may be associated with, at least in part, the platinum resistance in ovarian CCC. Thus, it would be reasonable to treat ovarian CCC using agents whose effects are not subject to NER repair.
2. Trabectedin
Trabectedin, an anticancer agent, has recently become the focus of attention for researchers investigating the treatment of ovarian cancer. Trabectedin is an antineoplastic agent that was originally derived from the Caribbean marine tunicate Ecteinascidia turbinata. It binds covalently to the minor groove of DNA, bending the DNA toward the major groove and disrupting its transcription, leading to G2-M cell-cycle arrest and ultimately apoptosis [55]. On the basis of the positive results of a phase III clinical study (OVA301 study) [56], the use of trabectedin in combination with pegylated liposomal doxorubicin was approved by the European Union in 2009 as a treatment for relapsed platinum-sensitive ovarian cancer.
Trabectedin interacts with the NER machinery in an unusual manner. An elegant study demonstrated that NER-deficient cells (deficient in NER-related genes) exhibited resistance to trabectedin and that their sensitivity to trabectedin was restored by the transfection of the corresponding genes [57]. These findings are in clear contrast with the results obtained for platinum-based agents. Moreover, interestingly, a previous study demonstrated that cells in the G1 phase are most sensitive to trabectedin [58]. As NER activity is increased in ovarian CCC cells and more than half of ovarian CCC cells are in G1-phase [59], trabectedin might be effective in patients with ovarian CCC. In preclinical investigations, we found that trabectedin shows the greatest in vitro antiproliferative effect than any other anticancer agents that are clinically used in the treatment of ovarian CCC [59]. We also found that the in vitro antiproliferative effect of trabectedin is further enhanced by the co-treatment with mTORC1 inhibitor [59]. Moreover, a strong synergy was observed when trabectedin was combined with SN-38, an active metabolite of irinotecan [60]. These results indicate that trabectedin plus mTORC1 inhibitor and/or irinotecan is worth investigating in the future clinical trials [60]. Currently, the clinical activity of trabectedin plus mTORC1 inhibitor everolimus is being evaluated in a phase II study involving recurrent ovarian CCC patients in Japan, and a preliminary evaluation showed that the drug exhibited significant activity, with a response rate of 14.3% and a clinical benefit rate of 42.9% [61].
3. Lurbinectedin
Lurbinectedin (PM01183) is a novel synthetic agent derived from trabectedin. It is also a covalent DNA minor groove binder and is structurally similar to trabectedin except for the replacement of a tetrahydroisoquinoline present in trabectedin by a tetrahydro β-carboline. This structural difference confers pharmacokinetic and pharmacodynamic benefits, which result in decreased toxicities and allow treatment regimens with increased dose intensity that lead to increased antitumor activity when compared to trabectedin [62]. In preclinical studies, Lurbinectedin exhibited broad antitumor activity against human cancer cell lines in vitro [63]. Lurbinectedin also significantly inhibited the growth of a wide variety of human cancer xenografts in athymic mice [62]. Following the encouraging results obtained in preclinical studies and phase I to II clinical trials [626364], a phase III trial investigating the activity of lurbinectedin versus pegylated liposomal doxorubicin or topotecan against recurrent ovarian cancer is currently being conducted [45]. We hope this agent be evaluated in the future clinical trials in patients with ovarian CCC.
CHEMOPREVENTION OF OVARIAN CCC
Chemoprevention is an ideal strategy to reduce the deaths from ovarian CCC as it may decrease the incidence or delay the tumor-onset. Recently, a mouse model of ovarian CCC carrying coexisting ARID1A and PIK3CA mutations has been developed [25], Moreover, the activated AKT/mTOR activity has been demonstrated in ovarian endometriosis, from which ovarian CCC is thought to arise [65]. We have previously demonstrated that the mTORC1 inhibitor (everolimus) markedly delayed tumor development and progression in a GEM that develops poorly differentiated ovarian cancer with activated AKT/mTOR signaling [66]. These results may indicate that PI3K/AKT/mTOR signaling is an important target for the chemoprevention of ovarian CCC, and that the potential of PI3K/AKT/mTOR inhibitors to prevent or delay the onset of ovarian CCC might be worth investigating. Recently, metformin, which have a potential to inhibit mTORC1, has attracted attention because of its chemopreventive activity [6768]. I hope that the chemopreventive agents such as PI3K/AKT/mTOR inhibitors or metformin be evaluated in women who are at high risk of developing ovarian CCC; i.e., women with ovarian endometriosis.
CONCLUSIONS
Ovarian CCC has specific biological and clinical behaviors, and the development of novel treatments based on its molecular characteristics is urgently needed. The PI3K/AKT/mTOR, VEGF, IL-6/STAT3, MET, and HNF-1β pathways are frequently activated and are promising therapeutic targets in ovarian CCC. On the basis of promising preclinical findings, some inhibitors targeting these pathways are currently being evaluated as treatment strategies for ovarian CCC, either monotherapy or in combination with other targeted/cytotoxic agents. Another treatment strategy which may improve the prognosis of CCC patients is the use of checkpoint inhibitors. According to recent reports, although the number of patients are very limited, nivolumab or avelumab showed antitumor efficacy in patients with CCC [6970]. Thus, the efficacy of immunotherapy in patients with CCC should also be investigated in the future clinical trial.
Given the potential toxicity of these treatments, it is important to identify biomarkers that can be used to predict which patients will benefit from such treatments. Moreover, a deeper understanding of the mechanisms responsible for chemoresistance, and the identification of effective treatment combinations will maximize the potential of these treatments. Furthermore, as a hierarchical cluster analysis using high-resolution microarray-based comparative genomic hybridization revealed that ovarian CCC constitutes a heterogeneous disease at the genomic level despite having similar histological features, and that the level of genetic complexity of CCC may predict the patient’s prognosis [71]. Thus, the molecular stratification may be crucial to optimize the therapeutic strategies for patients with CCC. Overcoming these challenges will aid the development of optimal therapies for ovarian CCC.
 XML Download
XML Download