

 PDF
PDF ePub
ePub Citation
Citation Print
Print


INTRODUCTION
Malignant peripheral nerve sheath tumor (MPNST) is a rare soft tissue sarcoma of ectomesenchimal origin. They can usually occur on schwann cell, peripheral nerve fiber and sheath and they are generally found on the extremities and the head and the neck. They are rarely found on the abdominal organ or cavity [1]. More than half of them occur related to neurofibromatosis type-I (NF-1), so the primary hepatic MPNST cases without NF-1 have been reported rarely [234]. To date, only 11 cases have been reported in the literature [2].
We herein describe a primary hepatic huge MPNST successfully treated with surgery, radiotherapy and adjuvant chemotherapy.
CASE REPORT
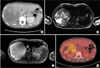
A 33-year-old female was admitted Soonchunhyang University Cheonan Hospital with a right flank pain for a weak. She underwent left ovarian cystectomy 8 years ago and had no other medical history. On physical examination, the abdomen was soft and flat with mild tenderness of the right flank area. And other physical examination was unremarkable with no skin lesion (i.e., café au lait). In laboratory findings, mild anemia, and mild prolonged coagulation profile were checked but liver function test and urinalysis were all within the normal limit. The serum tumor markers for α-FP, CEA, and CA 19-9 were also within the normal ranges. She was treated conservatively for several days but her symptoms did not subside and laboratory findings were still unremarkable. So we performed in radiologic investigation. The CT scan showed an approximately 12.5 × 11-cm sized, heterogenous enhancing mass in the right liver lobe (Fig. 1A). The mass showed hypodense in the precontrast phase and was contained a punctuate calcification and was not septated. MRI also showed a 13.0 × 11.0-cm sized, a heterogeneous huge mass in T1 weighted image, not washed out in portal enhancement phase. The mass was shown by heterogeneous high SI in T2WI and slightly high SI in hepatobiliary phase (Fig. 1B, C). There was a no fat suppression portion in definitely. An F-18 fluorodeoxyglucose PET (FDG-PET) showed huge mixed density mass with abnormal uptake in the lesion, with liver right lobe. The inner portion of mass was not filled by FDG and peripheral portion of mass was filled by FDG on max standardized uptake values 4.51 (Fig. 1D). Surgical exploration of the abdominal cavity revealed a 20.0 × 16.0-cm sized mass on the right hepatic lobe with attachment to right diaphragmatic pleura, so right hepatectomy and partial resection of adherent part to diaphragmatic pleura were done.
Histologically, the specimen was measured about 20.0 cm × 16.0 cm × 8.5 cm. The cut surface of the mass showed a round relatively demarcated solid pale gray mass (16.5 cm × 12.5 cm). Surgical resection margins were negative (Fig. 2A, B). The microscopic features were hyperchromatic spindle cells arranged in fasciculated pattern and abundant, faintly eosinophilic cytoplasm. The tumor cells showed microscopic invasion into the peritumoral hepatic tissue. Necrosis and hemorrhage was present. On fédération nationale des centres de lutte contre le cancer system, the tumor differentiation sore was 3, the mitotic count was 8 on 10 high power field, necrosis was 30% (Fig. 3A, B). On immunohistochemistry, the tumor cells were positive for s-100 protein, vimentin (Fig. 3C, D). The following immunohistochemical reactions were all negative: panCK, CK7, CK8/18, CK20, EMA, calretinin, SMA, myogenin, desmin, MyoD1, mdm2, CD99, CD10, CD31, CD34, hepatocyte Ab, glutamine synthetase, HSP70, glypican, c-kit (CD117), α-FP, ER, GFAP, HMB45, p63, TLE-1.
The patients were discharged at 13 days after operation without complications. In 6 weeks later, the patient was treated with adjuvant radiotherapy of 6,000 cGy for invaded side of diaphragm and was performed four courses of adjuvant chemotherapy with adriamycin, ifosfamide and cisplatin during a 6-month period. She is doing well without recurrence at 36-month follow-up after her first operation.
DISCUSSION
MPNST had been named malignant shwannoma in the past and is thought to originate from only schwann cell. But it was found that they also arise from a peripheral nerve sheath and fiber, fibroblast. So, World Health Organization in 1987 coined the term MPNST, replacing previous ambiguous terminologies such as malignant schwannoma, malignant neurofibrosarcoma, and malignant neurilemmoma [5].
The incidence of MPNST is 1 per 100,000 population and it constitutes between 3% to 10% of all soft tissue sarcomas. It is mostly found on upper and lower extremities and trunk. It's frequency is almost the same on male and female. These tumors may arise spontaneously in adult patients, although 5% to 42% of MPNST have an association with multiple NF-I. Peak incidence ages of MPNST are fourth decades on NF-1 patients and they have been known a main cause of death on NF-1 patients [1]. Primary hepatic MPNST is very rare with only 11 case reports in the literature. To our knowledge, our case is the 12th [234]. Nine of them are not associated with NF-1. In 4 cases, the patients expired within 4 months, in 1 case the patient died after 15 months after initial admission, and the other 6 cases were not followed up. Median age of diagnosis is 45, and male to female ratio is 9:3 and laboratory findings are usually nonspecific and radiologic findings demonstrate a heterogenous rapidly enlarging mass [34].
Histologic findings contain hyperchromatic spindle cell and abundant eosinophilic cytoplasm. However, in the majority of the cases, it is insufficient to confirm the diagnosis. Deferential diagnosis includes monophasic synovial sarcoma, leiomyosarcoma, fibrosarcoma [6]. Therefore, we also had to check and confirm its nervous tissue origin by immunohistochemical stains such as S-100 protein, vimentin, and TLE-1. S-100 protein is nonspecific but it has been known to be the strongest determination factor diagnosing MPNST [6]. Malignant severity of MPNST is related to the increasing mitotic count and the dysplasia of cells, the positive tumor necrosis and the infiltrating tumor margin [5].
Radical resection is the most important treatment of MPNST. And routine nodal dissection is not indicated [57]. Generally, MPNST have been considered chemotherapy and radiotherapy resistant tumor. So, it is difficult to define the role of adjuvant radiation and chemotherapy. But in the current series, better outcome was observed for advanced MPNST patients treated with doxorubicin-ifosfamide combination compared to other histological subtype soft tissue sarcoma patients [8]. In the past, the majority of the randomized chemotherapy trial had been shown no significant response [7]. But currently, on The Italian and German soft-tissue sarcoma cooperative group had been reported the overall response rate up to 45% in children group [9]. It have been reported that radiation therapy relates on lower local recurrence rate but does not reduce distant metastasis and overall survival rate. But recently, because of the effect on local recurrence control, it is recommended by International Consensus Statement as part of treatment, despite having no significant effect of long term survival rate [10].
Generally the prognosis of MPNST is very poor. 5-year survival rates have been reported about 30%–50%. Local recurrence rate and distant metastasis rate are very high after radical resection. The rates of distant metastasis have been reported 20%–50% on the average 16–22 months after surgery. The distant metastasis sites are lung, soft tissue, bone, liver, adrenal gland and peritoneal cavity. The tumor size, histological grade and resection margin have been known important prognostic factors. And other factors including age, location, with or without NF-1 are controversial [8]. Therefore in our case, we performed radiation therapy and chemotherapy after surgery and the patient is doing well without recurrence for 36 months.
In conclusion, this is the first case report about combination treatments including operation and adjuvant radiation and chemotherapy of primary hepatic MPNST without NF-1 patient. Our case is followed up for 3 years after operation without recurrence. Although MPNST is very rare and its prognosis is generally poor, it can be treated successfully with meticulous combined therapy.


 XML Download
XML Download