

 PDF
PDF ePub
ePub Citation
Citation Print
Print


INTRODUCTION
Currarino triad, also called Currarino syndrome, consisting of anorectal stenosis or other anorectal malformations, sacral bony anomalies and presacral mass, was first described by Currarino et al. [1] in 1981. This disease is related to the mutation of the MNX1 gene (motor neuron and pancreas homeobox 1, HGNC ID: 4979), previously called the HLXB9 gene. Mutations in the coding sequence of the MNX1 gene have been reported in nearly all familial Currarino triad cases and in approximately 30% of sporadic cases [2].
Cases of Currarino triad associated with Müllerian duct anomalies, such as duplication of the vagina and uterus, have already been reported [3]. However, inherited cases combined of both diseases without MNX1 gene mutation were rarely reported. We report familial cases; mother and daughter, having Currarino triad and Müllerian duct anomaly without MNX1 gene mutation; along with a brief review of the related literature.
CASE REPORTS
Case 1: Mother
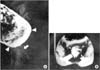
A 5-year-old girl was referred to our institution due to progressive constipation and incontinence since birth. Colon study showed a severe anorectal stenosis (Fig. 1A). CT myelogram revealed anterior meningocele that seemed to cause anorectal stenosis by compressing the rectum and sacral bony defect (Fig. 1B). She also had a horseshoe kidney. Ligation of sacral meningocele, removal of presacral lipoma, and anal dilatation with Hegar dilatators were performed. However, constipation did not improve, and reconducted colon study showed a persistent anorectal stenosis. Thus, posterior sagittal anorectoplasty was performed. The patient was discharged with improved defecation and continued to do well without additional treatment. When she was 27 years old, didelphic uterus was discovered at the work up for pregnancy. This patient was the first Currarino triad patient reported in Korea [4].
Case 2: Daughter
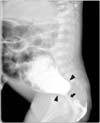
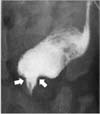
A newborn female was admitted due to vomiting, constipation and distended abdomen. The baby was born at 37 gestational weeks weighing 2.7 kg. Her mother, case 1, was impregnated naturally without assisted reproductive technology and denied any complications associating with pregnancy, including pregnancy-induced diabetes mellitus. The only problem during pregnancy was asymmetric shape of abdomen due to asymmetric enlargement of didelphic uterus. Meconium passage was not observed until 24 hours after birth. Colon study showed anorectal stenosis (Fig. 2). Pelvic magnetic resonance imaging revealed lobulating fatty mass in the presacral area, sacral bony defect and Müllerian duct didelphism (Fig. 3). Posterior sagittal anorectoplasty with sacrococcygeal teratoma excision was performed after diverting colostomy, and presacral mass was diagnosed as mature teratoma. Ganglion cells did not exist at the excised rectum on pathologic exam, but initially, we regarded the findings as a normally existing aganglionic segment at far distal rectum, not as Hirschsprung disease. Colon study was performed before colostomy repair to check up and evaluate the Hirschsprung disease. Transitional zone was suspected in the rectum (Fig. 4). Thus, Laparoscopy-assisted Soave operation was done under the impression of the Hirschsprung disease. Ganglion cells also did not exist at the distal part of the excised specimen. There was no sign of constipation, while intermittent soiling existed. At 25 months after Soave operation, at 34 months old, the patient did not show any symptoms of constipation but sometimes encountered occasional incontinence when she had loose stool, likely due to weak anal sphincter.
After diagnosis of both mother and daughter, evaluating the MNX1 gene mutations was attempted. The gene was extracted from peripheral blood samples and evaluated by performing PCR and sequencing reaction at the department of laboratory medicine, Seoul National University Hospital. The mutations were not detected in both mother and daughter. They denied having any relatives with Currarino triad or Hirschsprung disease.
DISCUSSION
After first the report of Currarino triad in 1981, more than 300 cases have been reported until now. The majority of the reports were descriptions of sporadic and isolated cases [5]. But, in some case series, familial cases were found and Yates et al. suggested the disease had familial tendencies and showed autosomal dominant inheritance [67]. In 1998, the disease causing gene was reported as HLXB9, now called MNX1 [8]. The MNX1 gene locates at human chromosome 7q36 and codes for a 403 amino acid protein that consists of a highly conserved 82 amino acid domain, a homeo-domain and a poly-alanine region. This protein plays important roles in pancreatic development and motor neuron differentiation in the spinal cord [6]. MNX1 mutations were found in nearly all familial Currarino triad cases [2].
Currarino triad has fairly variable presentations and many additional associated malformations have been described. Especially, Currarino triad combined with Müllerian duct anomalies have been reported in up to 15% of cases [3], whereas, the incidence of Müllerian duct anomalies in the general population is about 7% [9]. Müllerian duct anomalies mainly occur sporadically and most familial cases are multifactorial. Other modes of inheritance, including autosomal dominant, autosomal recessive, and X-linked disorders, also exist [10]. Müllerian duplications may be observed in Currarino triad patients, and a correlation with the malformation sequences of Currarino triad has to be confirmed [3].
Currarino triad has various phenotypes. Within family members who had same MNX1 gene mutation, the presentations were different, even patients who revealed near normal phenotype with the same mutation were reported. In addition, caudal dysgenesis syndrome, heterogeneous association of congenital caudal anomalies affecting the caudal spine and spinal cord, the hindgut, the urogenital system and the lower limbs, also existed. This disease is known having similar phenotype with Currarino triad but previous studies did not prove the relationship with MNX1 gene mutations [26]. MNX1 gene mutation may not be the only cause of Currarino triad, as other causes may exist.
Environmental factors, genetic heterogeneity and somatic mosaicism could be considered as possible explanations for familial Currarino triad patients without MNX1 mutations and some of the sporadic nonmutated patients. Unknown modified genes able to change the expression of contiguous genes, or located elsewhere in the genome, or due to epigenetic factors that can interfere with the MNX1 pathway, from the transcription to the protein activation, influence the severity of the deriving phenotypes [26]. They also might be a possible explanation.
In summary, though nearly all familial Currarino triad cases have reported having MNX1, these patients, mother and daughter who had Currarino triad with Müllerian duct didelphism, do not have relation to MNX1 gene mutation. It is true that MNX1 gene mutations are important for Currarino triad, but other genetic or environmental influencing factors could exist. Thus, we should be aware of this possibility for further research.

 XML Download
XML Download