

 PDF
PDF ePub
ePub Citation
Citation Print
Print


INTRODUCTION
Peutz-Jeghers syndrome (PJS) is a rare autosomal dominant inherited syndrome consisting of gastrointestinal hamartomatous polyps and mucocutaneous hyperpigmentation of the lips, buccal mucosa, and digits. Polyps can occur anywhere in the gastrointestinal tract and can grow large enough to cause bowel obstructions. The most common location for polyps is the small bowel (64%), although the colon (53%), stomach (49%), and rectum (32%) can be involved. Usually there are fewer than 20 polyps present in each case, varying in size from several millimeters to more than 5-cm diameter. Those polyps are present from childhood and may sometimes lead to intussusception or gastrointestinal bleeding [1].
The diagnostic criteria for PJS include the presence of characteristic muco-cutaneous pigmentation, small bowel hamartomatous polyps and family history of PJS. Patients need to fulfill two of these three criteria for the diagnosis.
Mutations in the serine-threonine kinase 11 (STK11/LKB1) gene on chromosome 19p13.3 are considered the major cause of PJS [2]. Germline mutations of STK11 are documented in up to 70%-80% of patients with PJS and up to 15% of cases have deletions of all or part of STK11 [3].
To our knowledge, this is a rarely reported case of PJS with germline mutation of STK11 in Korea. Therefore, we report this successfully treated case of PJS with a brief review of the relevant literature.
CASE REPORT
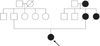
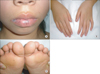
A 12-year-old young girl was admitted to emergency services with a long history of lower abdominal pain that had become more severe over the last 2 days, a mild iron-deficiency anemia and loss of appetite. The patient was not receiving any specific medication and her medical history did not suggest any major disease. There was no history of diarrhea, constipation, vomiting and/or gastrointestinal bleeding, but family history was significant for death from ovarian cancer at 41 years for maternal grandmother, death from colon cancer at 32 years for her mother, and colon cancer at 34 years for maternal aunt (Fig. 1). Physical examination revealed no abdominal tenderness, masses, or organomegaly, except for the presence of dark-brown pigmented maculae on the lower lip and melanin pigmentation spots on the hands and feet (Fig. 2). Laboratory check-up was also within the normal range except for mild anemia, with microcytosis and iron depletion, and stool was positive for occult blood. The abdominal x-ray examination showed no signs of acute abdomen. Preoperative abdominal CT showed two target-appearance lesions with mild proximal dilated small intestine and polyps.
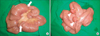
As laparotomy was indicated for the persistent abdominal pain and intussusception, we decided to perform exploratory laparotomy. After a full examination of abdominal organs and the small intestine, two jejuno-jejunal intussusceptions invaginated over large two polyps were found upon laparotomy, and after the intussusceptions were manually reduced, the polyps were removed by enterotomy (Fig. 3). Histopathological examination revealed that all polyps were hamartomatous in nature.
After obtaining informed consent, blood samples were collected from the patient for STK 11 gene analysis. RNA was extracted with blood RNA extraction kit (Genolution pharmaceuticals, Seoul, Korea). The whole nine exons of STK11 gene were amplified with 7 sets of primers (Table 1) using ImProm II Reverse Transcriptase (Promega, Madison, WI, USA) and h-Taq DNA polymerase system (Solgent, Daejeon, Korea). The amplified products were electrophoresed on the 1.5% agarose to confirm a single product, then analyzed with Denaturing High-Performance Liquid Chromatography (DHPLC) system (Transgenomics, Omaha, NE, USA). The unmatched chromatogram on the DHPLC was confirmed with ABI prism DNA analyzer (Applied Biosystems, Foster, CA, USA). In the chromatogram of sequence analysis, the patient revealed heterozygous missense mutation of codon 303 (c.303C > G, I303M, Fig. 4).
The patient recovered uneventfully and was discharged on the 8th postoperative day. Postoperative colonoscopy revealed 3 more pedunculated polyps, which were excised by snare polypectomy, but no mass lesion was seen. Histopathological examination of polyps revealed the hamartomatous nature. She was subsequently enrolled for cancer screening and remains well and asymptomatic at 26 months postsurgery.
DISCUSSION
PJS was first described in 1921 by Peutz and subsequently elaborated upon by Jeghers in 1949 [4]. It is an inherited, autosomal dominant disorder with variable inheritance, characterized by hamartomatous polyps in the gastrointestinal tract, mostly in the small bowel, and pigmented mucocutaneous lesions. Its incidence is calculated at 1 in 200,000 live borns, its mean age of onset is 25.2 years, and one-third of cases are diagnosed before the patient is 10 years old [4].
The presenting complaints of PJS are intestinal obstruction (43%), abdominal pain (23%), blood in the stool (14%), and anal extrusion of polyp (7%). The remaining 13% of cases are diagnosed because of melanin pigmentation. The most frequent complication in young age is intussusception, occurring in 47% of patients, primarily in the small intestine (in 95% of cases) [1]. Because multiple hamartomatous polyps could develop as multiple intussusceptions, the small bowel should be fully examined to be certain additional intussusceptions are not present. Our case of PJS developed two jejuno-jejunal intussusceptions invaginated over two large polyps that were found upon laparotomy.
There are three criteria for the diagnosis of PJS was established in 1997 (Tomlinson and Houston), 2000 (World Health Organization, WHO), 2010 (Mayo Clinic) (Table 2) [5]. The WHO criteria makes the point of stating the PJS pigmentation should be "prominent" as perioral pigmentation is common in the general population. The authors of Mayo Clinic have found that PJS pigmentation often fades after puberty and is sometimes very faint or even absent in adults and therefore have not included that in their criteria. The more recently developed Mayo criteria also differs from the WHO criteria by including LKB1 mutation testing. Our case would be diagnosed with PJS, since all three criteria were satisfied.
Mutations in the serine-threonine kinase 11 (STK11/LKB1) gene have been considered to be the major cause of PJS. The gene is divided into nine exons that encode a 433 amino acid protein that acts as a tumour suppressor. Mutation detection rates of 10%-94% have been achieved at different centers, depending on the screening method [6]. Most mutations are frameshift or nonsense changes, resulting in an abnormal truncated protein and the loss of kinase activity. The splice-junction alterations, insertions, and nucleotide or whole gene deletions have also been reported. However, the STK11 mutation spectrum and genotype-phenotype correlation are still poorly understood, but individuals with missense mutations had a significantly later time of onset to first polypectomy and other cancer symptoms compared with individuals with truncating mutations or no detectable mutations [7]. In this study, although the patient revealed a heterozygous missense mutation and remained well and asymptomatic, the regular follow-up should involve both the most efficient and least invasive procedures depending on available resources, the individual patient's disease manifestations, psychosocial situation, and personal preferences. It is necessary to perform consistent screening for possible malignancies in all patients with PJS: colonoscopy, upper endoscopy, CT, MRI or ultrasound of the pancreas, chest x-ray, mammography and pelvic examination with ultrasound in women, testicular examination in men, CA 19-9, and CA 125 [6].
In PJS with the STK mutation, the risk of developing cancer by age 70 years was 81%. The most common cancers identified were gastrointestinal in origin (esophagus, stomach, small bowel, colorectum, pancreas). The cumulative risk for these cancers at age 60 years was 42%. In women the risk of breast cancer was substantially increased to 32% by age 60 years [8]. If genetic testing is performed and a mutation is found in an affected family member, then genetic testing of at-risk relatives will provide true positive or negative test results. At-risk members who receive true negative test results have a risk of PJS similar to that of the general population. At-risk relatives who test positive should follow the surveillance guidelines. Genetic screening of young patients at high risk can be justified by the benefit of avoiding the higher morbidity of emergent versus elective laparotomy for small bowel obstruction. Individuals with STK11 mutation are at risk for a wide variety of cancers at a young age. Although there were no controlled studies on the effectiveness of cancer surveillance for PJS, a surveillance recommendation by age and sex has been developed from expert opinion [1]. This patient was enrolled for cancer screening and fortunately remains well and symptomatic at 26 months postsurgery.
Usually, polyps in the stomach or colon that are greater than 1 cm in size noted during endoscopic surveillance is recommeded polypectomy. Surgery has been recommended for symptomatic or rapidly growing small intestinal polyps or asymptomatic polyps greater than 1-1.5 cm in size. Some experts suggest that an attempt to clear the small intestine of polyps ("clean sweep") should be made during laparotomy. This can be facilitated by concomitant interoperative endoscopy with polypectomy or, in the case of larger polyps, enterotomy. The clean sweep approach appears to decrease the need for recurrent small bowel surgery [9]. Double balloon enteroscopy for removal of small bowel PJS polyps has been reported and might decrease the need for laparotomy. In our case, postoperative colonoscopy was performed by surveillance guidelines, and revealed 3 more pedunculated, 0.5-cm-sized polyps, which were excised by snare polypectomy.
Therefore, we report this rare case in the hope that although extremely rare, the small bowel should be fully examined to be certain additional intussusceptions are not present in PJS patients at laparotomy, and for emphasizing the important role of genetic testing for definite diagnosis, appropriate genetic counseling, and surveillance guidelines.



 XML Download
XML Download