

 ePub
ePub Citation
Citation Print
Print


INTRODUCTION
Pheochromocytomas or paragangliomas are rare neuroendocrine tumor that arise from chromaffin cells. The tumor produces catecholamines, such as norepinephrine and epinephrine, and leads to well-known clinical symptoms, such as hypertension, headache, sweating, palpitation, and orthostatic hypertension.
A pheochromocytoma or a paraganglioma that exclusively secretes dopamine is extremely rare. It is usually found at extra-adrenal sites and is diagnosed as a subtype of paraganglioma. The high dopamine level in this tumor is caused by a deficiency in dopamine-beta-hydroxylase, which converts dopamine to norepinephrine [1]. For this reason, this tumor differs from classic pheochromocytomas in many respects, not only in its clinical features but also in its oncologic aspect. Only two original articles and five case reports have reported the occurrence of dopamine secreting paragangliomas in the retroperitoneum around the adrenal glands [2-8].
We experienced the first case in Korea in an asymptomatic woman. We report this interesting case and establish clinical concepts for this rare tumor by reviewing the related literature.
CASE REPORT
A 26-year-old Korean woman was referred after the accidental detection of an adrenal mass in her right side. She denied any medical history including endocrine disorders. There was no family history of endocrine disease. She had no hypertension, palpitation, sweating, headache, flank pain, or mass upon palpation of her abdomen.
On physical examination, her blood pressure was 110/90 mmHg and her heart rate was 90 beats/min. The results of laboratory tests, including a complete blood count and liver and renal function tests, were all unremarkable. The results of an overnight 1 mg dexamethasone suppression test were normal. Serum norepinephrine and epinephrine levels were normal but the dopamine level was high (425 ng/L). Analysis of a 24-hour urine sample revealed marked elevation of the urinary dopamine level (1,565.3 µg/day) but normal levels of vanillylmandelic acid (VMA), norepinephrine, epinephrine, metanephrine and normetanephrine.
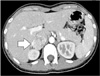
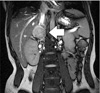
Computed tomography results showed a 4.3 × 3.2 cm hypervascular mass on the right adrenal gland with an early washout enhancement pattern. The mass abutted against the inferior vena cava (IVC) (Fig. 1). Magnetic resonance imaging revealed a 2.8 cm sized tumor that was located in the right periadrenal area and that abutted against the adrenal gland laterally and IVC medially (Fig. 2).
Our preoperative diagnosis was a dopamine producing paraganglioma in the right retroperitoneum. She was not administered a preoperative α-blocker or any anti-hypertensive drug. Hand-assisted laparoscopic right adrenalectomy was performed because of the firm attachment of the tumor to adjacent structures. En-bloc excision of the tumor and adrenal gland was performed (Fig. 3). Total operative time was 110 minutes and blood loss was minimal. Her blood pressure was continuously normal and there were no fluctuations in vital signs during the operation. She was discharged with no immediate postoperative complications.
On the histologic report, the tumor measured 3.4 × 3.0 × 2.5 cm. It was well encapsulated, homogenous and had a yellowish cut-surface with some internal hemorrhagic spots. Microscopic diagnosis was a neuroendocrine tumor whose clinical manifestations were consistent with those of a paraganglioma. Hematoxylin/eosin staining revealed vascular invasion and extension to peritumoral adipose tissue. An immunohistochemical study showed that the tumor was positive for neuroendocrine markers such as CD-56, synaptophysin and chromogranin. The tumor had a Ki-67 index of less than 1 percent and was negative for S-100 (Fig. 4).
Her 24-hour urine dopamine level returned to normal (388.4 µg/day) after the operation. Her general condition was good and there was no recurrence during a 10-month follow-up.
DISCUSSION
Among the many types of paraganglioma, an exclusively dopamine producing type is extremely rare. Only seven cases are listed in the worldwide database and no case has ever before been reported in South Korea [2-8].
The exclusive production of dopamine can be ascribed to a deficiency in dopamine β-hydroxylase which converts dopamine to norepinephrine in the catecholamine biosynthesis pathway beginning with tyrosine [1,4,8]. Because of the abnormal catecholamine synthesis pathway, the levels of dopamine and its end product, homovanillic acid (HVA), are increased, whereas the levels of norepinephrine, epinephrine and their end product, VMA, are decreased. In consequence, the clinical presentation of this tumor is different from that of a classical pheochromocytoma that secretes norepinephrine and epinephrine. There is an absence of hemodynamic changes or so-called paroxysmal symptoms that are usually found in patients with catecholamine secreting pheochromocytoma. As a result, the patients with exclusively dopamine producing paraganglioma are mostly asymptomatic at initial diagnosis. Some constitutional symptoms such as fever, malaise, weight loss or diarrhea may occur as a result of the increased circulating dopamine level. Because paragangliomas tend to grow larger than adrenal pheochromocytomas, some patients experience vague abdominal pain or experience a palpable abdominal mass [4-8]. The asymptomatic features make it difficult for clinicians to detect the tumor at an early stage. Detection is even more challenging to physicians working in institutes without routine screening procedures for the detection of catecholamine and dopamine.
Differential diagnosis can be achieved by measuring the dopamine levels in serum and 24-hour urine samples. Clinicians should not rule out the possibility of this rare tumor when evaluating adrenal masses with normal serum norepinephrine, epinephrine and urinary VMA levels, even if the radiologic images strongly suggest a pheochromocytoma.
The treatment of every adrenal tumor should involve complete surgical excision. Laparoscopic surgery is considered the standard surgical option. Hand-assisted laparoscopic surgery has been adapted for the removal of large tumors and for the rescue of operative procedures in complicated cases of laparoscopic surgery [9].
To minimize surgical mortality caused by hypertensive crisis during the manipulation of the tumor, preoperative administration of a α-blocker is essential for norepinephrine and epinephrine producing pheochromocytomas. However, α-blocker treatment is contraindicated for exclusively dopamine secreting paraganglioma because it can cause profound cardiovascular collapse after surgery, and can even result in death following a hypotensive crisis [8]. The explanation for this fatal outcome is as follows. First, unlike norepinephrine and epinephrine, dopamine has no affinity for the α-receptor. Second, it has been suggested that dopamine can lower blood pressure by itself. There are two subtypes of dopamine receptors; the D1 receptor, which relaxes smooth muscles in blood vessels, and the D2 receptor, which inhibits noradrenaline release from postganglionic sympathetic neurons. These two effects could explain the pre-operative hypotension in dopamine secreting tumor patients. Blood pressure is sometimes elevated after the removal of this tumor presumably because of the same mechanism [4,5].
Dopamine secreting paragangliomas are more likely to present malignant features than classic catecholamine secreting type paragangliomas or pheochromocytomas. This rare tumor tends to be diagnosed in patients who are asymptomatic, and the tumor is detected later than tumors that secrete norepinepnrine, epinephrine or both. Delayed diagnosis may be related to the higher incidence of malignant transformation and metastasis. Concerning the biochemical features, one report suggests that the decreased activity of dopamine β-hydroxylase results in poor differentiation of this tumor, which may explain why the dopamine-secreting type shows advanced malignant features [10]. There are known risk factors for judging the malignant potential of paragangliomas. Elevated dopamine or HVA levels in 24-hour urine (>120 nmol/g) is correlated with large tumor size and malignant potential. Extra-adrenal location, a tumor weight of more than 80 g, DNA aneuploidy or triploidy and persistent postoperative hypertension have also been reported as risk factors for malignant pheochromocytoma [10]. Because of the rarity of this tumor, there are no definite criteria for evaluating its malignant potential.
In conclusion, exclusively dopamine secreting paragangliomas are extremely rare. Lack of clinical symptoms and the rarity of this tumor can lead to the lack of, or a delay in diagnosis. In addition to radiologic imaging, dopamine assays in serum and 24-hour urine collection are useful diagnostic tests. Laparoscopic tumor removal is the treatment of choice without administration of a preoperative α-blocker. Prognosis is known to be poorer than that for pheochromocytomas.


 XML Download
XML Download