

 ePub
ePub Citation
Citation Print
Print


INTRODUCTION
Pheochromocytoma was well known as "10 percent tumor". It is widely believed that approximately 10% of pheochromocytomas are associated with familial syndromes. However, it is now recognized that the frequency of germline mutations in apparently sporadic pheochromocytoma is as high as 24% [1]. Hereditary pheochromocytoma could be a phenotype of multiple endocrine neoplasia (MEN) type 2, Von Hipple-Lindau disease, familial pheochromocytoma-paraganglioma and neurofibromatosis type 1. The causal genes were discovered as RET, VHL, SDHB, SDHD and NF1 respectively. Each genetic syndromes share common characteristics such as early onset, multifocality and bilaterality. So in case of youngaged, bilateral pheochromocytoma, searching for genetic mutation is recommended [2]. We report a case of a 44-year-old man with bilateral pheochromocytomas whose genetic study shows a rare germline L790F mutation of RET oncogene.
CASE REPORT
A 44-year-old man was admitted for incidentally found bilateral bulky adrenal mass lesions. He daily drank one or two bottles of Soju (Korean distilled spirits). A computed tomography (CT) scan was taken for evaluation of alcoholic liver disease. He felt paroxysmal attack of palpitation and sweating since last year, but had no medication. His systolic blood pressure was 110 mmHg and diastolic blood pressure was 65 mmHg. His heart rate was 70 per minute. On the physical examination, no mass was palpable in abdomen and neck lesion.
In the hormonal studies, 24 hours urinary metanephrine and vanillylmandelic acid was elevated as 6.12 mg/day (normal value, less than 1.3 mg/day) and 23.38 mg/day (normal value, less than 8 mg/day). Twenty-four hours urinary cortisol was normal as 45.9 ug/day (normal value, 20 to 90 ug/day). Plasma rennin activity and aldosterone level was also normal as 0.44 ng/mL/hr (normal value, 0.15 to 2.33 ng/mL/hr) and 65.0 pg/mL (normal value, 35.7 to 240.0 pg/mL).
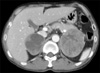
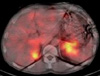
On the CT scan, right adrenal mass was 6 cm sized, well defined and mostly necrotic. The left adrenal mass was 8 cm, hypervascular and centrally necrotic (Fig. 1). I-123 metaiodobenzylguanidine scan showed bilateral adrenal uptake with no evidence of metastasis (Fig. 2).
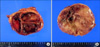
The patient was diagnosed as bilateral pheochromocytomas. After prescribing alpha-blocker terazosin for twelve days, bilateral adrenalectomy was done under general anesthesia. On the operation, both adrenal masses were well marginated without invasion and lymph node metastasis (Fig. 3). The pathological diagnosis was also pheochromocytoma. He was recovered uneventfully, discharged with gluco-corticoid and mineralo-corticoid replacement.
We tested germline mutation of RET oncogene with patient's informed consent. Germline mutation was analyzed by direct sequencing of exons 10, 11, 13, 14, 15, 16 of RET oncogene. At the codon 790 of exon 13, missense mutation of L790F was found. It was a point mutation of DNA change from TTG to TTT, resulted amino acid change from Leucine to Phenylalanine (Fig. 4).
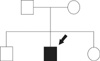
After the result of germline mutation of RET oncogene, we diagnosed the patient as multiple endocrine neoplasia type 2A, and searched an evidence of medullary thyroid cancer in the patient and his family members. In his family members, there was no history of thyroid cancer or endocrine diseases (Fig. 5). We checked an ultrasonography of patient's thyroid, but there was no evidence of thyroid nodule. The patient's calcitonin level was 3.7 pg/mL (normal value, less than 20 pg/mL). We checked the stimulated calcitonin level, but the result was also normal range. We explained the results and recommended prophylactic total thyroidectomy and genetic test about his parents, but he rejected. So we made a plan to check the calcitonin and thyroid ultrasonography regularly.
DISCUSSION
About 10 percent of pheochromocytomas were assumed to be hereditary. However, recent advances of genetic studies show much higher germline mutation rates than that of previous reports. According to the European Network for the Study of Adrenal Tumors Pheochromocytoma Working Group [2], germline mutation rate was 25.9% from 642 pheochromocytoma and paraganglioma patients. In the other study [1], 24.3% of the 271 sporadic pheochromocytoma patients had germline mutations. It is now estimated that about 20 to 30% of pheochromocytomas have hereditary tendency.
Hereditary pheochromocytoma is a phenotype of five genes; VHL, RET, SDHB, SDHD and NF1. From the reports of the European Network [2], each mutation rates were 8.7% in VHL gene, 5.3% in SDHB, 4.8% in SDHD, 4.8% in RET and 3.7% in NF1 respectively. Other European study [1] shows similar results; 11.1% in VHL, 4.4% in SDHB, 4.1% in SDHD, 4.8% in RET.
Each genetic syndrome has characteristic phenotypes. It is possible to anticipate genotype by characteristic phenotype, so identification of clinical characteristics is very important. Genetic testing for NF1 gene is not routinely carried out. Diagnosis of neurofibromatosis type 1 is possible based on the typical clinical features; café-au-lait spots, neurofibromas and axillary and inguinal freckling. On the contrary, genetic test of NF1 gene is difficult and costly due to large sized NF1 gene, composed of 57 exons without hot spots. In this case we can exclude NF1 mutation based on the clinical features.
Von Hippel-Lindau disease is characterized by hemangioblastomas, renal tumors, pancreatic and endolymphatic sac tumors. Pheochromocytomas occur in about 26% of von Hippel-Lindau patients [3]. Associated pheochromocytoma is typically lack of phenylethanolamine-N-methyltransferase which converts noradrenaline to adrenaline, resulted in high normetanephrine and normal metanephrine [4]. In this case, we can rule out VHL mutation due to high level of metanephrine.
SDHB and SDHD gene are recently identified as paraganglioma syndrome type 4 and type 1. These genes are subunits of the mitochondrial complex II, which is involved in the Krebs cycle as succinate dehydrogenase. Pheochromocytoma with SDHB and SDHD germline mutation is frequently malignant, extra-adrenal or bilateral. So in that phenotype, genetic test about SDHB and SDHD gene are necessary [2].
Germline mutation of RET oncogene is associated with MEN type 2 which has typical character of genotype-phenotype correlation and mutation hot spots. Medullary thyroid cancer is the most frequent and malignant tumor in MEN type 2. MEN type 2 was classified according to the aggressiveness and onset of medullary thyroid cancer. The penetrance of medullary thyroid cancer is different according to the mutation site. Patients with level 1 mutations (codons 609, 768, 790, 791, 804 and 891) have the lowest risk for medullary thyroid cancer, patients with level 2 mutations (codons 611, 618, 620 and 634) are intermediate risk, and patient with level 3 mutations (codons 883 and 918) have the highest risk for medullary thyroid cancer [5]. Medullary thyroid cancer was developed in 100% of level 3, 73% of level 2 and only 45% of level 1 RET gene mutation [6].
In this case, L790F mutation was found in RET oncogene, which was not reported before in Korea. 1998, Berndt et al. [7] first described a new hot spot for mutations affecting codon 790 of RET oncogene. They reported that nine (69%) of 13 carriers with L790F mutation had developed medullary thyroid cancer. Initially, L790F mutation was reported to be associated with pheochromocytoma, but in the following study [8], L790F mutation rarely associated with pheochromocytoma.
Interestingly, this case shows bilateral pheochromocytomas which was rare phenotype of L790F and does not show medullary thyroid cancer which was common phenotype of L790F. Machens et al. [6] reported that mean diagnostic age of pheochromocytoma was 46.5 years and medullary thyroid cancer was 51.6 years in level 1 RET mutation. We can expect patient's occurrence of medullary thyroid cancer in the near future.
Like as risk classification of medullary thyroid cancer, risk of pheochromocytoma also can be classified. The highest-risk category of pheochromocytoma includes SDHB, SDHD and the level 3 risk of RET; the high-risk category includes VHL missense mutation and the level 2 risk of RET; the least-high risk category includes VHL truncating mutations and level 1 risk of RET [9].
The incidence of germline mutation of multifocal or bilateral pheochromocytomas was much higher than that of unilateral pheochromocytoma. In the 26 patients of multifocal or bilateral pheochromocytomas, 80% of them had a mutation (46% in VHL, 19% in RET, 15% in SDHD, none in SDHB). In the other study [10], 12 RET, 1 VHL, 1 SDHD gene mutations were found in the 23 bilateral pheochromocytomas. They recommended sequential mutational analysis of RET, followed by VHL and SDHD in bilateral pheochromocytoma.
This case is the first report of L790F RET germline mutation in Korea. In case of bilateral pheochromocytoma, germline mutation test for hereditary pheochromocytoma is necessary.

 XML Download
XML Download