

 ePub
ePub Citation
Citation Print
Print


INTRODUCTION
Type 1 neurofibromatosis patients (NF1) have a significant predisposition to GISTs [1]. Ras activation through the somatic inactivation of the NF1 gene seems to be involved in the pathogenesis of NF1-associated GISTs [2]. Contrary to sporadic GISTs, the NF1-associated GISTs are characterized by a large number of small-sized tumors, typical location in the small intestine, and rare mutations of the KIT or PDGFRA [1]. However, occasional mutations of both the KIT and the PDGFRA have been reported in the NF1-associated GISTs, and the meaning of these mutations remains unclear [3-6]. We present a case of NF1-associated GISTs with a missense point mutation of the KIT in a 52-year old woman.
CASE REPORT
Clinical summary
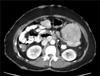
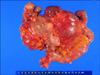
A 52-year-old woman with a history of NF1 presented with intermittent abdominal pain. She had been diagnosed as NF1 30 years ago and had a family history of NF1. Her skin showed numerous café au lait spots and multiple cutaneous neurofibromas over the whole body. A physical examination demonstrated a 20 × 15 cm palpable mass in the left abdomen. The abdominal computed tomography revealed a 15 × 9 × 7 cm tumor and several smaller masses along the jejunal loop (Fig. 1). On laparotomy, main tumor and numerous (more than 50) small nodular masses in the jejunum, located over 60 cm of the Treitz ligamen were found (Fig. 2). Segmental resection of the jejunum was performed and the patient recovered uneventfully.
Pathologic findings
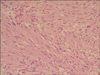
The largest tumor was 18 × 11 × 6 cm, showed mainly extramural growth and connected to the ulcerative mass at the mucosal side. Focal hemorrhagic and necrotic portions were seen at the cut surface. Some of the multiple nodular masses, ranging in size from 0.2 to 4.0 cm, showed intraluminal umbilication. Histologically, the tumors consisted of interlacing fascicles of uniform spindle cells with eosinophilic cytoplasm and elongated nuclei (Fig. 3). The largest tumor had increased mitoses (19/50 high power fields) whereas the mitotic count of smaller tumors was less than 5 per 50 high power fields. Immunohistochemical staining revealed tumor cells positive for CD117, CD34 and Vimentin, and negative for S-100 and smooth muscle actin(Fig. 4).
The mutation analysis of KIT and PDGFRA
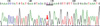
Genomic DNA was extracted from each paraffin blocks of two tumors, including the extramural and intramural portions of the largest one. Exons 9, 11, 13 and 17 of KIT and exons 12 and 18 of PDGFRA were amplified using the polymerase chain reaction (PCR) and then directly sequenced. The extramural portion of the largest tumor harbored a missense point mutation (Trp557Gly) of the KIT exon 11 (Fig. 5). The intramural portion of the largest tumor, as well as the other tumor, had wild type KIT and PDGFRA.
DISCUSSION
Gastrointestinal stromal tumors (GISTs) are the most common mesenchymal tumors of the gastrointestinal tract, with characteristic morphologic, immunophenotypic and molecular features [7]. The gain of function KIT and PDGFRA mutations are considered to be a major driving force in the pathogenesis of sporadic, non-familial GISTs [8,9]. Activating mutations in the KIT gene are present in up to 90% of the GISTs, and 35% of the GISTs lacking KIT mutations have activation mutations in the related receptor tyrosinekinase gene, PDGFRA. Most of the KIT mutations are located on the following exons: 11 (the juxtamembrane domain), 9 (the extracellular domain), 13 (the TK I domain) and 17 (the TK II domain); conversely, the PDGFRA mutations are usually located on the exons 12 or 18. The KIT-JM domain encoded by the exon 11 is the most common mutational "hot spot" in GISTs [8]. A great majority of the KIT exon 11 mutations are deletion/deletion-insertion and missense mutations. They represent the second most common type of KIT exon 11 mutations found in GISTs. These mutations cluster on the 5' KIT exon 11 and almost exclusively involve the KIT codons 557, 559 and 560. Normally KIT and PDGFRA are activated by their ligands, stem cell factors and PDGFs. Ligand binding to the receptor EC domain results in dimerization of the receptors and phosphorylation of the tyrosines in their cytoplasmic TK domains. This leads to a phosphorylation cascade of the tyrosine residues in multiple downstream signaling molecules and to activation of the signal transduction pathways, including the Ras/MAP kinase signaling network. These KIT and PDGFRA mutations lead to ligand independent dimerization of the receptors and downstream the phosphorylation cascade, ultimately leading to activation of cellular proliferation.
In western countries, type 1 neurofibromatosis is a common autosomal dominant inherited disorder, with a prevalence of one in 3,000 live births. The syndrome results from either inherited or spontaneous germ-line mutations in the NF1 gene located at chromosome 17q11.2, which encodes the tumor suppressor gene, neurofibromin. In addition to multiple cutaneous and deep-seated neurofibromas, patients with NF1 develop other evidence of the disease, including skin manifestations (e.g., axillary freckling, café au lait spots), neurologic disorders, extra-intestinal neoplasm and neoplasm of the gastrointestinal tract (e.g., ampullary adenocarcinomas, somatostatinomas, and GISTs) [5].
The association between GISTs and type 1 neurofibromatosis has been well established. The clinico-pathologic characteristics of the GISTs in NF1 are different from those of sporadic cases; most cases show multiple GISTs, predominantly involving the small intestine, and mutations of the KIT or PDGFRA are rare [1]. Neurofibromin, the protein encoded by the NF1 gene, functions as a GTPase-activating protein for Ras by catalyzing the hydrolysis of the active Ras-GTP to an inactive Ras-GDP. Additional somatic inactivation of the wild type NF1 allele in the ICCs seems to be the molecular event underlying the GIST development in the NF1 setting [2]. Because of these genetic disorders, patients with NF1 are estimated to develop GISTs at a rate of at least 45 times higher than sporadic cases and GISTs are considered as a type of the clinical symptoms seen in NF1.
Contrary to the sporadic GISTs, only occasional KIT and PDGFRA mutations have been reported in the NF1-associated GISTs. Ten KIT/PDGFRA mutations have been reported in the 87 investigated patients (Table 1). Takazawa et al. [3] identified two KIT (e.g., Pro627Leu and Ile654Thr) and two PDGFRA (e.g., Pro589Lys and Arg822Ser) missense mutations in two separate tumors of each patient. These mutations do not correspond to the 'GIST-type' of mutations and might therefore be random genetic events related to the tumor progression. Five 'GIST type' of mutations were found in the NF1-associated GISTs. Yantiss at al. [5] found a point mutation (Val559Asp) of the KIT exon 11 in three tumors from a patient. However, the presence of identical mutations in separated tumors raises the possibility of a germline mutation. Additionally, four mutations were identified in two other studies [4,6], i.e., two in-frame deletions (Trp557_Lys558del and Val560del) of the exon 11 and one duplication (Ala502_Tyr503dup) of the exon 9 of KIT and one PDGFRA mutation (Asp842Val) in the exon 18. One missense mutation (Trp557Gly) of the KIT exon 11 was identified in the extramural portion of the largest tumor found in the presented case. We consider that this is a 'GIST type' mutation. Both the intramural portion of the largest tumor and the other tumor had wild type KIT and PDGFRA. The mutational feature of the presented case suggests that this mutation is not the initiation event, but might be an acquired genetic event which occurred at the 'hot spot' of the KIT mutation in the late phase of the development of NF1-associated GISTs.
Imatinib mesylate, a selective inhibitor of both KIT and PDGFRA, blocks these constitutively activated tyrosine kinases. This agent is currently being used as a molecular target drug for treating the GISTs and is showing a remarkable therapeutic effect on most of the GISTs [7]. Most of the NF1-associated GISTs lack the KIT and PDGFRA mutation, therefore imatinib mesylate is not expected to be effective on these multiple tumors. However, there was one report demonstrating the therapeutic effect of imatinib mesylate on the NF1-associated GIST with wild type KIT and PDGFRA [10]. Data on the effect of tyrosine kinase inhibitors on the NF1-associated GISTs with activating mutations are, nevertheless, limited.
Molecular analysis for more cases is needed to understand the meaning of occasional occurrences of either KIT or PDGFRA mutations and to characterize their clinical features, including the response to tyrosine kinase inhibitors of the NF1-associated GISTs, according to the presence of the KIT or PDGFRA mutations.


 XML Download
XML Download