

 Citation
Citation Print
Print


INTRODUCTION
Atherosclerosis is understood recently to be an inflammatory process that involves the infiltration of inflammatory cells and complex structures that consists of smooth muscle cells, macrophages, lymphocytes, microvessels and different types of collagen [1-4]. It is known that the major cell types of inflammatory cells are macrophages and T-lymphocytes which are closely associated with plaque rupture [5]. In addition, vascular smooth muscle cells (VSMCs) proliferation has been identified in early atherosclerotic lesions. This is in contrast to the low level proliferation of VSMCs detected in advanced lesions, which suggests that the VSMCs might be senescent, as demonstrated by the senescence-associated β-galactosidase (SAβG) activity [1,2,6].
As the atherosclerosis progresses, the VSMCs become a senescent and undergo the apoptosis [7,8]. The apoptotic cells were found in areas near or within the lipid rich core, and the apoptosis of VSMCs is associated with the remodeling and stability of atherosclerotic plaque [9,10]. In critical regions of the plaque, the apoptosis of VSMCs predisposes the plaque to rupture and subsequent thrombosis [11].
The interaction between apoptotic VSMCs and inflammatory cells, such as macrophages and T-lymphocytes, leads to the thinning of the fibrous cap overlying the lipid-rich core and this contributes to plaque rupture. It has been known that apolipoprotein (APO) C1 and APO E are expressed in macrophages and most of the expressed genes induced by macrophage colony-stimulating factor were identified to be APO C1 and APO E [12,13]. Further, the expression of APO C1 is highly correlated with an APO E expression [12]. The expressed macrophage and T-cells may affect the degradation of the extracellular matrix and the depletion of VSMCs, which are both seen in the development of atherosclerosis. We have previously reported that APO C1 and APO E were prominently expressed in carotid atherosclerotic lesions, as compared to non-atherosclerotic arteries [14].
In the present study, we compared the regional differences of the expressions and senescence of cellular components, such as VSMCs, cluster of differentiation 68 (CD68, macrophage), CD3 (T-lymphocyte), APO CI, and APO E, in atherosclerotic lesions. In addition, we identified that the expression of cellular components in less advanced atherosclerotic plaques by comparing more advanced atherosclerotic plaques.
METHODS
Specimens
A total of 10 atherosclerotic tissues were obtained (arteries from carotid endarterectomy) and compared with five non-atherosclerotic tissues as a control group (arteries from brain death donors). The tissue specimens were immediately frozen and stored at -70℃ in liquid nitrogen. This study was approved by the Institutional Review Board at our institution and written informed consent was obtained from the patients to use their tissues. The Investigation conforms with the principles outlined in the Declaration of Helsinki.
Western blot analysis
Fifty µg protein from each specimen was used for the analyses of human telomerase reverse transcriptase (hTERT, diluted 1:1,000; Santa Cruz Biotechnology, Santa Cruz, CA, USA), CD68, SM-α actin, APO C1, APO E, and actin by performing Sodium dodecyl sulfate-polyacrylamide gel electrophoresis on 8% to 15% gels. The protein samples were then incubated with polyclonal rabbit anti-human hTERT, monoclonal mouse anti-human CD68 (diluted 1:100; Dako, Glostrup, Denmark), monoclonal mouse anti-human α-smooth muscle actin (SM-α actin, diluted 1:400; Dako), polyclonal rabbit anti-human apolipoprotein C1 (diluted 1:200; Chmi-con, Rosemont, IL, USA), polyclonal rabbit anti-human apolipoprotein E (diluted 1:500; Dako) and polyclonal goat anti-human actin (diluted 1:200; Santa Cruz Biotechnology) as primary antibodies for 1 hour and next they were incubated with polyclonal goat anti-rabbit immunoglobulin/horseradish peroxidase (HRP) (diluted 1:2,000; Dako), polyclonal rabbit anti-goat immunoglobulin/HRP (diluted 1:2,000; Dako) and monoclonal goat anti-mouse immunoglobulin/HRP (diluted 1:2,000; Becton, Dickinson and Company, Franklin Lakes, NJ, USA) as secondary antibodies during 1 hour. Following that, the specimens were transferred onto hybond enhanced chemiluminescence reagents (ECL; Amersham Canada, Oakville, ON, Canada) nitrocellulose membranes (Schleicher and Schuell, Keene, NH, USA) and these were finally developed with the use of ECL.
SAβG staining
The SAβG activity was determined by using a SAβG staining kit from Cell Signaling Technology (Beverly, MA, USA). The tissues were washed twice in phosphate-buffered saline (PBS), fixed for 15 minutes at room temperature in fixative solution (2% formaldehyde, 0.2% glutaraldehyde), washed twice in PBS, and next incubated for 24 hours at 37℃ in SAβG staining solution (pH 6.0) that contained 1 mg/mL 5-bromo-4-chrolo-3-indlyl β-D-galactopylanoside, 5 mmol/L potassium ferrocyanide, 5 mmol/L potassium fericyanide, 150 mmol/L NaCl, 2 mmol/L MgCl2, 0.01% sodium deoxycholate and 0.02% Nonidet-40. The 10 µm-thick formalin-fixed, paraffin-embedded sections were then subjected to immunohistochemistry.
Immunohistochemistry
After deparaffinization followed by dehydration, the serial paraformaldehyde-fixed paraffin sections were rinsed 3 times for 5 minutes each time in PBS. The paraffin-embedded tissues were blocked with using Dako Cytomation peroxidase blocking reagent buffer for 30 minutes. The slides were covered with goat serum for 1 hour and then incubated with the following antibodies: monoclonal mouse anti-human α-smooth muscle actin (diluted 1:50; Dako), monoclonal mouse anti-human CD68 (diluted 1:100; Dako) and, polyclonal rabbit anti-human CD3 (diluted 1:50; Dako) for 1 hour at room temperature, and then, the slides were washed for 3 times for 5 minutes each time in PBS. The slides were then exposed to envision buffer for 30 minutes and this was followed by treatment with diaminobenzidine for 30 minutes. Finally the slides were dehydrated in ethanol and xylene.
RESULTS
Expression of inflammatory cells and apolipoproteins in the main and adjacent lesions of atherosclerotic plaques
Cellular senescence and inflammatory cells in the shoulder area of atherosclerotic plaques
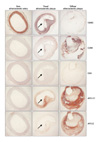
β-galactosidase (β-gal) activity, which is known as a senescence marker of atherosclerosis, was represented in the carotid atherosclerotic plaque (green area in Fig. 2A-2) but not in the non-atherosclerotic artery (Fig. 2A-1). In addition, VSMCs, macrophages and T-lymphocytes were expressed in the media of the β-gal stained areas (Fig. 2B-3-1, 4-1, and 5-1). The shoulder lesions, indicated as boxes in Fig. 2B-3-1, 4-1, and 5-1, had a small number of VSMCs as compared with the number of macrophages and T-lymphocytes. The expression of T-lymphocytes was more prominent in the β-gal stained area, as compared with the non-stained area (Fig. 2B-4, 4-1).
Expression of inflammatory cells and apolipoproteins in the 'focal' and 'diffuse' atherosclerotic plaques
The sites of critical stenosis were retrieved from the endarterectomy specimens. Atherosclerotic plaques in critical stenosis were divided into 'focal' and 'diffuse' atherosclerotic plaques according to the morphological features. The focal atherosclerotic plaques showed small pools of extracellular lipid and hyperplasia of the VSMCs and diffuse atherosclerotic plaques demonstrated a rich necrotic core and/or a core of extracellular lipid, including cholesterol crystals. The diffuse atherosclerotic plaques were considered to be a more advanced atherosclerotic lesion than the focal plaques because these had a larger area of the plaque and a more prominent expression of inflammatory cells and only the rare expression of the VSMCs (Fig. 3). The histological examination revealed that the number of VSMCs was higher in the focal atherosclerotic plaques than that in the non-atherosclerotic artery (Fig. 3). Macrophages and T-lymphocytes expressed in the focal and diffuse atherosclerotic lesions, but not in the non-atherosclerotic arteries. In addition, the expression of inflammatory cells was more prominent in the shoulder area of the focal lesions (arrow in Fig. 3). The expressions of APO C1 and APO E were showed in the focal and the diffuse lesions, but not in the non-atherosclerotic arteries (Fig. 3). In the diffuse lesion, macrophages were prominently expressed in the fibrous cap. Moreover, APO E was mainly represented in the regions around the necrotic core and fibrous cap, and APO C1 was mainly demonstrated within the necrotic core.
DISCUSSION
The development of atherosclerosis is induced by the migration, proliferation and differentiation of inflammatory cells, mainly macrophages and T-lymphocytes. The monocytes differentiate into macrophages in the vessel wall and the macrophages produce various proteases that degrade the extracellular matrix [15]. Our study found that there was a high expression of macrophages in the main lesion of atherosclerotic plaques as compared with that of the area adjacent to the main lesion, as was assessed by Western blot analysis (Fig. 1A). Macrophages express APO E, which has an anti-proliferative effect on VSMCs [16]. APO E null mice have been extensively used as an animal model for atherosclerosis as these mice develop atherosclerosis that is similar to that found in human [17,18]. In addition, it has been previously reported that hTERT-APO E doubly deficient mice show impaired proliferation of both lymphocytes and macrophages, and reduced atherosclerosis [19]. However, we found that APO E and hTERT were more highly expressed in atherosclerotic lesions than in non-atherosclerotic arteries (Fig. 1A). This finding suggested that the expressions of APO E and hTERT in atherosclerotic plaque may be affected by the proliferation of macrophages during the development of atherosclerosis. Also, we suppose that the difference of expression for these proteins in a same atherosclerotic patient may be the important causes to the presence of atherosclerotic lesion.
The expression of APO C1 is highly correlated with the APO E expression, and this is probably the result of their close location in the APO E/C1/C4/C2 gene cluster [8]. We found that APO C1 and APO E were overexpressed in atherosclerotic lesions, as compared with that of non-atherosclerotic arteries, by performing Western blot analysis (Fig. 1A).
Foam cells of a macrophage origin, as indicated by immunoreactivity for CD-68, kill the adjacent VSMCs in the atherosclerotic plaque and the rupture sites are also characterized by the paucity of VSMCs and the high concentration of macrophages and inflammatory cells [20-22]. Our histologic examinations indicated that there were high concentrations of macrophages and T-lymphocytes and the loss of VSMCs in the shoulder area and this area is known to be prone to plaque rupture [9]. This study, we showed that the loss of VSMCs and the prominent expression of macrophages during the progression of atherosclerosis (Fig. 3).
β-gal staining is a useful marker for cellular senescence in both in vitro and in vivo because β-galactosidase is a metabolic enzyme that is highly expressed in senescent cells. But, there is still a need to identify whether the cells showing β-gal staining are genuine senescent cells because β-gal positive cells may be macrophages with a high lysosomal content. Interestingly, our study demonstrated that most of the areas showing β-gal staining exactly overlapped with the areas that expressed T-lymphocytes in atherosclerotic plaques, as compared with macrophages and VSMCs. Therefore, β-gal staining in cells is possibly not due to a high lysosomal content, but rather, this is due to cellular senescence. It indicates that the T-lymphocytes may be a crucial feature in atherosclerosis and it is also closely associated with the progression of atherosclerosis.
Macrophages and T-lymphocytes are known to be major cell types that found at sites of rupture [7] and, in the present study, the senescence of these cells and the lack of VSMCs were observed in the shoulder area of atherosclerotic plaques (Fig. 2). Our findings showed that large lipid-rich necrotic core were present in the 'diffuse' atherosclerotic lesions and this is resulted of increased apoptosis and decreased scavenging, which characterizes more advanced atherosclerotic plaque. APO C1 and APO E were highly expressed in the 'focal' and 'diffuse' atherosclerotic plaques, as compared with that in the non-atherosclerotic arteries. In particular, macrophages were prominently in the fibrous cap, APO C1 was mainly expressed within the necrotic core and APO E was particularly expressed around the necrotic core and fibrous cap in more advanced atherosclerotic plaques (Fig. 3). These findings suggest that APO C1 and APO E are strongly associated with the expansion of necrotic core during the progression of atherosclerosis.
In conclusion, our findings represent that expression and senescence of macrophage and T-cells involved in the progression of atherosclerosis and suggest that expression and deposition of APO C1 and APO E contributes to the destabilization of atherosclerotic plaque by expanding the necrotic core. Therefore, the inhibition of macrophage, T-cells, the APO C1 and APO E may well become an increasingly important therapeutic strategy for preventing the progression of atherosclerosis.


 XML Download
XML Download