

 PDF
PDF ePub
ePub Citation
Citation Print
Print


TO THE EDITOR: In acute lymphoblastic leukemia, t(9;22) occurs more commonly in adults (25–30%) than in children (3%) and its incidence increases exponentially with age [1]. This translocation confers a poor prognosis to patients and results in high white blood cell count in addition to high incidence of central nervous system involvement and splenomegaly. Additional cytogenetic aberrations occur in 60% of cases of Philadelphia positive adult acute lymphoblastic leukemia, of which monosomy 7 and 9p abnormalities are considered to be the most common [2].
On the other hand, t(17;19)(q22;p13) is a very rare translocation in acute lymphoblastic leukemia that is associated with an extremely poor prognosis. Coexistence of t(17;19)(q22;p13) with Philadelphia chromosome has only been reported once in the literature [3].
Case
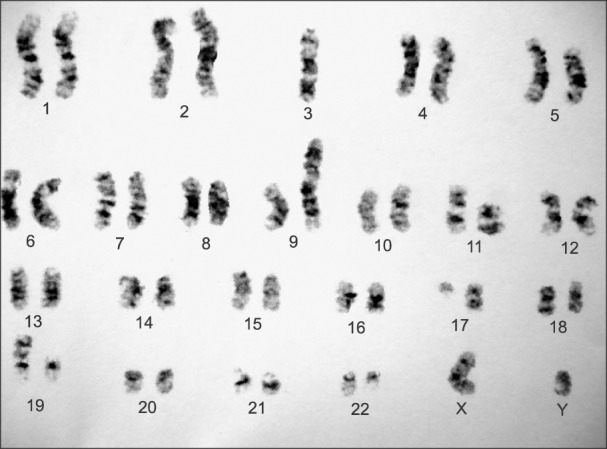
A 41-year-old man was admitted to hospital due to bone pain and abdominal discomfort. Pallor and splenomegaly were the only two abnormal findings on physical examination. Laboratory tests showed prominent leukocytosis, anemia and thrombocytopenia (white blood cell count=170,000/µL, hemoglobin=9.2 g/dL, platelet count= 30,000/µL). Flow cytometry of bone marrow aspiration revealed 80% blast cells with B cell precursor phenotype expressing HLA-DR, CD19, CD10, CD22, tdt, CD20. Bone marrow cytogenetic study with Giemsa banding method exhibited 45XY t(9;22) (q34;q11) de(3) der(9) t(3;9)(q11;q13) del3(q10) t(17;19)(q11;p13) (Fig. 1). Reverse transcription polymerase chain reaction was performed on a bone marrow specimen which also revealed m-bcr-abl1 fusion gene (p 190).
During hospitalization, the patient had an episode of epistaxis and his coagulation tests showed prolonged prothrombin time (PT=19.2 sec, normal range, 11–13 sec), partial thromboplastin time (PTT=42.5 sec, normal range, 25–35 sec) and low fibrinogen level (fibrinogen=92 mg/dL, normal range, 200–400 mg/dL). Thus, he underwent treatment with fresh frozen plasma till the symptoms resolved and the fibrinogen level reached beyond 100 mg/dL. He also received induction chemotherapy with Hyper CVAD chemotherapy regimen including cycles A and B. During cycle A, he took 1) cyclophosphamide with Mesna 500 mg IV on the first 3 days, 2) vincristine 2 mg IV on days 4 and 11, 3) adryamycin 50 mg on day 4, and 4) dexamethasone 40 mg on days 1 to 4 and 11 to 14. Three weeks later cycle B was initiated with the following combination: 1) methotrexate IV infusion 1,600 mg on the first day, 2) folinic acid 30 mg every 6 hours for 8 doses, 3) cytarabine 3,000 mg every 12 hours for 4 doses on 2nd and 3rd days, and 3) methylprednisolone 30 mg twice daily for 6 doses on days 1, 2 and 3. Imatinib (600 mg daily) was also added to his regimen after the detection of Philadelphia chromosome. Meanwhile, the patient received 12 mg intrathecal methotrexate weekly. Unfortunately, the patient's peripheral smear showed prominent leukocytosis (white blood cell count=140,000/µL) with 80% blast cells after completion of hyper-CVAD regimen (four courses of cycle A and B). Then he became candidate for bone marrow transplantation after salvage chemotherapy but he refused to receive salvage chemotherapy regimen. Therefore, there was no remedy but to continue treatment with 6-mercaptopurine (50 mg three times daily), methotrexate (15 mg weekly), vincristine (2 mg monthly) and another tyrosine kinase inhibitor, nilotinib (400 mg daily). Subsequent peripheral blood smear showed no blast cells and complete blood count yielded the following: white blood cells=2,500/µL, hemoglobin=10 g/dL and platelets=25,000/µL.
Discussion
The Philadelphia chromosome was the first chromosomal aberration found to be associated with malignancy [4]. In fact, it is a diminutive chromosome 22 derived from t(9;22)(q34;q11). This translocation can result in three differently sized fusion products: p190, p210 and rarely p230. This fusion gene has a tyrosine kinase activity and occurs in chronic myelogenous leukemia, acute biphenotypic leukemia, and acute lymphoblastic leukemia. It has recently been discovered that an intragenic deletion of the IKZF1 gene discriminates Philadelphia positive acute lymphoblastic leukemia patients from other t(9;22)(q34;q11) cases [5]. Additional chromosome aberrations are found in 40–86% of Philadelphia positive acute lymphoblastic leukemia cases, of which hyperdiploid karyotype, monosomy 7 and 9p abnormalities are the most common [26]. The incidence of t(9;22)(q34;q11) increases exponentially with age in acute lymphoblastic leukemia patients from 2% in children to 20–40% in adults. This chromosomal abnormality leads to leukocytosis, splenomegaly and central nervous system involvement. Dismal prognosis of this translocation has been improved after the advent of tyrosine kinase inhibitors [78].
t(17;19)(q22;p13) is an extremely rare translocation with an estimated incidence of 0.1% [4]. Among 9,000 acute lymphoblastic leukemia trial patients in the Leukemia Research Cytogenetic Group Survey, only 9 had t(17;19) (q22;p13). Their ages ranged from 5 to 18 years with a median age of 13 years. Acute lymphoblastic leukemia patients had a B-cell precursor phenotype and none of them had remarkable leukocytosis (50×109/L) [9]. It is supposed to be a variant of t(1;19)(q23;p13) and results in fusion of the HLF gene to TCF3 [10]. At the molecular level, there are two different breakpoint positions for this translocation. In type 1, chimeric oncoprotein arises from breakpoint within intron 13 of TCF3 and intron 3 HLF. This type of translocation is associated with disseminated intravascular coagulation. The pathogenesis of hemostatic system activation in acute leukemia is probably due to tumor cell procoagulant activities, fibrinolytic properties and cytokine release [1112]. In type 2, breakpoints in intron 12 of TCF3 and intron 3 of HLF rearrange and make fusion oncoprotein which is associated with hypercalcemia via parathyroid hormone related protein mediation [12]. This case revealed a different breakpoint on the long arm of chromosome 17 in q11 instead of its usual site q22 and seems to be a very rare type of t(17;19) which has been reported once in a case of acute lymphoblastic lymphoma [13].
In a recent review of the literature, 21 cases with t(17;19)(q22;p13) were reported and their clinical presentation and outcome were evaluated. They were all children and teenagers with ages ranging from 3 to 16 years, and there was a slight female predominance. Nine cases showed evidence of disseminated intravascular coagulation at diagnosis or during the course of the disease. Twelve cases also demonstrated hypercalcemia in their laboratory tests. Almost all of them died due to relapse of leukemia or post-chemotherapy infection [14].
Our case had some features of both t(9;22) and t(17;19). The present case was an adult who had remarkable leukocytosis and splenomegaly which are all common in Philadelphia positive acute lymphoblastic leukemia and rare in t(17;19). On the other hand, he also had disseminated intravascular coagulation, which is a feature of t(17;19). We reviewed the literature and found only one case of Philadelphia positive adult lymphoblastic leukemia with t(17;19) whose clinical presentation was relatively similar to ours. That case was a 44-year-old woman with weakness, splenomegaly and leukocytosis [3]. These findings show that coincidence of t(17;19) and t(9;22) tends to be seen in adults rather than children and may be associated with remarkable leukocytosis and splenomegaly in addition to disseminated intravascular coagulation or hypercalcemia. Glover et al. [15] exhibited in vitro susceptibility of leukemic blasts from a patient to tyrosine kinase inhibitors. However, its effectiveness is not improved in vivo due to the rarity of cases with t(17;19).
 XML Download
XML Download