

 PDF
PDF ePub
ePub Citation
Citation Print
Print


TO THE EDITOR: Amyloidosis of the gastrointestinal tract is uncommon and may lead to malabsorption. A 62-year-old woman visited our hematology clinic with a 3-month history of dysphagia and easy fatigability. She felt more difficulties in swallowing solid food than liquid food and easy fatigue on daily activities without orthopnea or paroxysmal nocturnal dyspnea. She felt no fever, anorexia, night sweats, weight loss, or lump in the neck. In addition, the patient had well controlled hypertension and rheumatoid arthritis with oral medications.
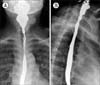
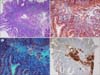
The blood pressure was 130/80 mmHg and the pulse rate was 98 beats/minute. On physical examination, marked pallor, glossitis and angular cheilitis were found. The barium swallow test revealed esophageal stricture with an anterior shelf-like projection at the hypopharynx and cervical esophagus junction, suggestive of cricopharyngeal web (Fig. 1). The complete blood counts analysis showed hemoglobin level of 6.0 g/dL, white cells counts of 4.5×109/L, and platelet counts of 700×109/L. On peripheral blood smear, microcytic hypochromic red cells with aniso-poikilocytosis were remarkable with some pencil cells. The iron profile (serum iron, 31 mg/dL; iron-binding capacity, 435 mg/dL; transferrin saturation, 7%; and serum ferritin, 10 ng/mL) was consistent with iron deficiency anemia (IDA). The results of fecal occult blood, IgA-tissue transglutaminase antibody, anti-nuclear antibody, and urea breath test for Helicobacter pylori were negative. No parasites/cyst/ova were detectable on stool examination. Although an esophagogastroduodenoscopy (EGD) was performed to evaluate the etiology of IDA, no remarkable finding was detected except for luminal narrowing at the level of cervical esophagus. Duodenal biopsy revealed a moderate lymphoplasmacytic infiltration in the lamina propria along with deposition of amyloid along blood vessels (Fig. 2). In the immunohistochemistry (IHC), serum amyloid A protein staining was positive without kappa/lambda light-chain restriction and transthyretin (TTR) staining was negative. No evidences of dysplasia, malignancy, or amyloid deposition were found on esophageal and duodenal biopsies. Serum/urine protein electrophoresis and immunofixation electrophoresis did not reveal any monoclonal band and serum free light-chain assay result was normal (Kappa, 1.2 mg/L; Lambda, 1.33 mg/L; K: λ, 0.9). In the bone marrow examination, plasma cells accounted for 4% of all nucleated cells, but amyloid deposits and light-chain restriction were negative in the IHC. Systemic workup for secondary amyloidosis including echocardiogram, abdominal ultrasonography, rectal biopsy, and colonoscopy (with biopsy) were negative for amyloid deposits. As a result, the patient was diagnosed as isolated duodenal amyloidosis (secondary) with Plummer-Vinson syndrome. The parenteral iron supplement (ferrous carboxymaltose at 900 mg total dose intravenously for 3 days) improved blood counts within 4 weeks (hemoglobin, 12.0 g/dL; white cell count, 5.5×109/L; and platelet count, 400×109/L) and led to complete resolution of stomatitis and glossitis with symptomatic relief of dysphagia. The patient has undergone regular follow-up with periodic (3 monthly) blood counts and liver/renal function monitoring.
Amyloidosis is defined as deposition of amyloid (an amorphous homogenous fibrillar material produced as a consequence of protein misfolding and identified by its characteristic apple green birefringence on polarized light microscopy by using Congo red stain and beta-pleated appearance on electron microscopy) in the extracellular space, causing distortion of tissue architecture and function [1]. Amyloidosis can be either systemic (the site of protein production is remote from its deposition) or localized (protein is produced at the site of deposition, with the usual sites being the respiratory tract, bladder, breast, skin, or gastrointestinal tract), the latter being a less common entity [23]. Systemic amyloidosis can be classified as amyloid light-chain (AL) type, amyloid A (AA) type, or familial transthyretin-associated (ATTR) type [4]. The symptoms are various depending on the organs involved. Because many gastrointestinal (GI) symptoms such as diarrhea, early satiety, and weight loss could be caused by other mechanisms unrelated to amyloidosis, the diagnosis of GI amyloidosis requires a biopsy-proven amyloid deposition in the GI tract [56].
GI amyloidosis is uncommon and is usually observed in systemic amyloidosis (7% of AL amyloidosis) [1]. Of the 2,334 patients with amyloidosis evaluated by Cowan et al. [6], 76 (3.3%) had biopsy-proven GI amyloidosis, which was systemic in 60 patients (2.57%) and localized in 16 (0.68%). Inayat and Hurairah [7] reported a case of localized duodenal amyloidosis (secondary, AA deposition) presenting as unexplained IDA that improved with iron supplementation. Given the fact that rheumatoid arthritis is the most common cause of AA amyloidosis, the etiology of our case was chronic inflammation resulting from rheumatoid arthritis [8]. As no evidence of GI bleeding was found on EGD, malabsorption was likely responsible for IDA.
In our opinion, presentation of localized GI amyloidosis (AA type) as Plummer-Vinson syndrome is exceptionally rare. This case highlights the importance of endoscopy in the initial workup of unexplained IDA and need for a biopsy even if no mucosal abnormality is identified. In addition, the differential diagnosis with systemic amyloidosis or plasma cell dyscrasia is also emphasized here, because localized amyloidosis usually could be controlled with conservative management.
 XML Download
XML Download