

 PDF
PDF ePub
ePub Citation
Citation Print
Print


INTRODUCTION
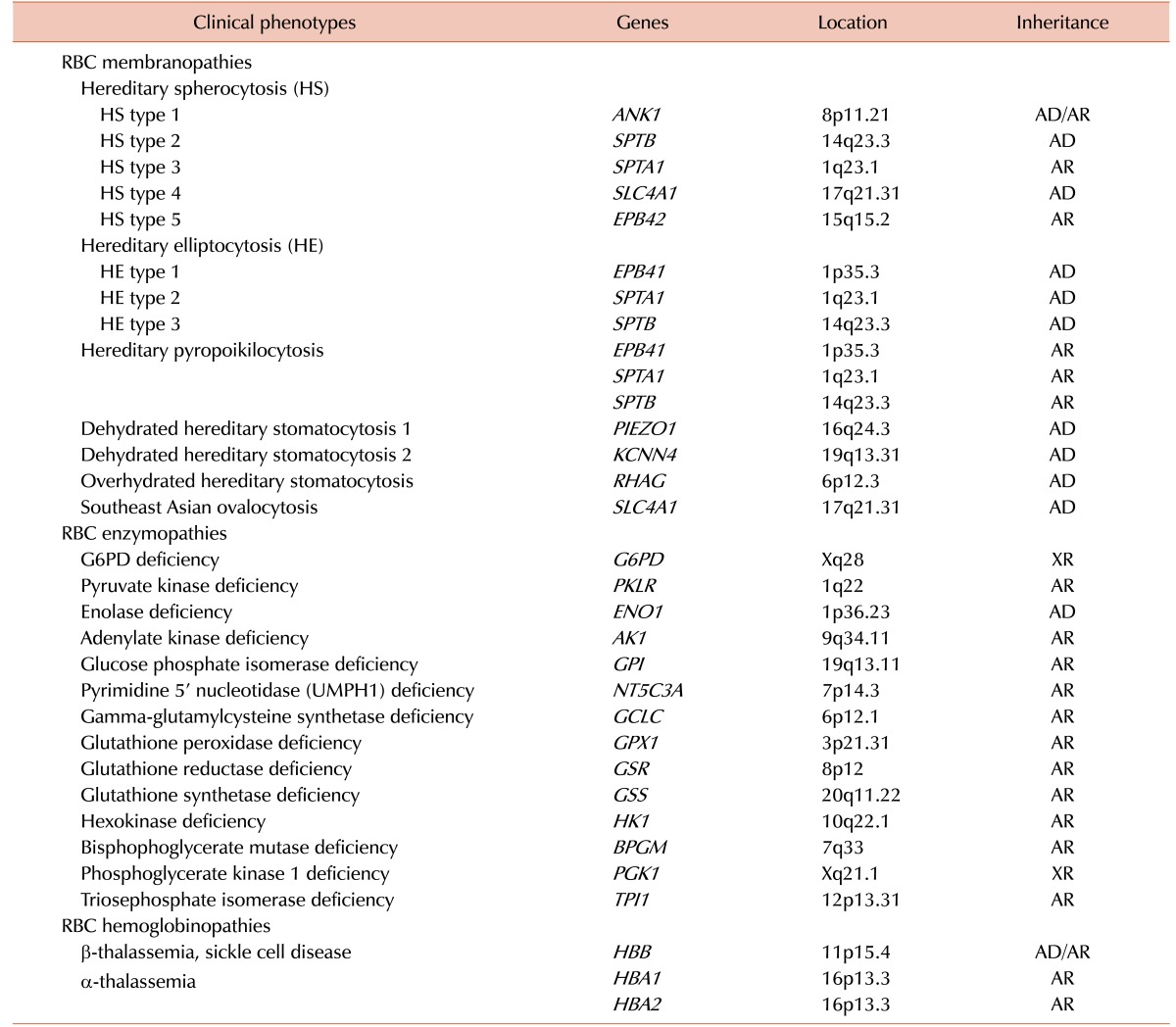
The term inherited hemolytic anemia (IHA) encompasses a diverse group of genetically and phenotypically heterogeneous disorders that result from an increase in the rate of RBC destruction [1]. The severity of the anemia or the course of the onset of hemolysis depends on the extent of this destruction. Mild hemolysis can be asymptomatic, while the anemia in severe hemolysis can be life threatening and cause angina and cardiopulmonary decompensation [23]. IHA develops when the RBCs themselves are defective (intrinsic HA). Defects in hemoglobin (Hb), the RBC membrane, and RBC enzymes are the major causes of intrinsic HA, which are commonly referred to as hemoglobinopathy, membranopathy, and enzymopathy, respectively [456]. Most intrinsic HAs are inherited disorders, but paroxysmal nocturnal hemoglobinuria is exceptional [7]. The most common of these disorders are α- and β-hemoglobinopathies, glucose-6-phosphate dehydrogenase (G6PD) deficiency, and hereditary spherocytosis (HS), which affect millions of people worldwide [8]. A Korean IHA survey-based study reported RBC membranopathies in 64.0%, hemoglobinopathies in 19.9%, and RBC enzymopathies in 13.3% of cases [9]. Traditional diagnosis of IHA has been done via a step-wise process including RBC morphology, membrane protein analysis, Hb electrophoresis, and measurement of RBC enzyme levels [10]. Occasionally, accurate diagnosis of IHA is a challenge because the clinical features may overlap in cases with different etiologies and it is not possible to distinguish between them using conventional diagnostic techniques [1112]. Therefore, there is still a need for more sensitive and reliable diagnostic tests to improve the accuracy of IHA diagnosis. A genome-wide association study of Hb concentration and related parameters (Hb, mean cell hemoglobin [MCH], mean cell hemoglobin concentration [MCHC], mean cell volume [MCV], packed cell volume [PCV], and RBC) has revealed the possible effects of 75 independent genetic loci on the genetic mechanisms and biological pathways that control RBC formation and function [13]. The range of genetic abnormalities has been extensively characterized [14] and genetic testing is available to identify the specific mutation(s) of IHAs (Table 1). Sanger sequencing is widely applied to identify disease-causing mutations in cases where traditional testing has failed or when a patient has been extensively transfused, leading to confounding biochemical and other testing findings due to mixed RBC populations. Recent technological advances including next-generation sequencing (NGS) provide a cost-effective and rapid approach to molecular diagnosis of IHA through extensive and simultaneous evaluation of a group of disease-causing genes [15].
This review describes effective diagnosis of IHA in the current genetic era and introduces recent insights into advanced genotype-phenotype correlation on the basis of genetics; we also discuss the possible implications of NGS in clinical practice for IHA patients.
RBC MEMBRANOPATHY
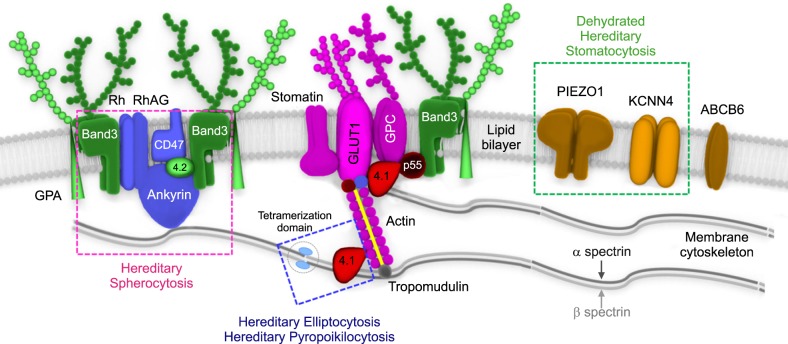
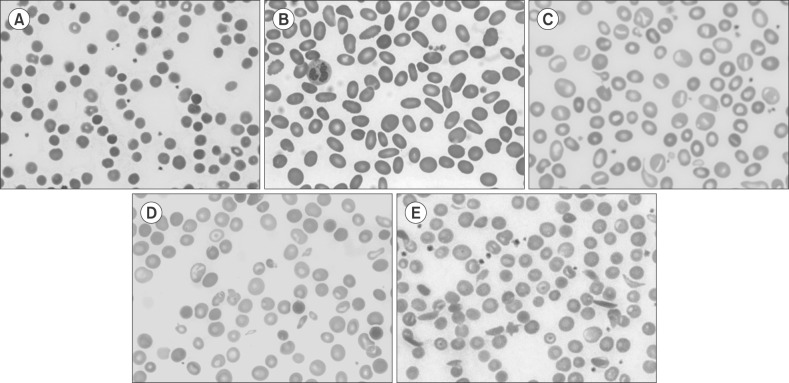
Knowledge of RBC membrane structure is important because defects in its structure underlie multiple IHAs [16]. The human RBC membrane consists of three basic components: a lipid bilayer, transmembrane linker proteins, and a two-dimensional spectrin-based cytoskeleton network [171819]. Connections of the two layers depend on different linker proteins with binding sites, respectively, for the cytoplasmic domains of the integral membrane proteins (band 3 and glycophorin C) embedded in the lipid bilayer and specific regions of spectrin proteins in the cytoskeleton (Fig. 1). RBC membranopathies are the result of qualitative abnormalities or quantitative deficiencies of the RBC cytoskeletal proteins and can be divided into those resulting from structural protein loss including HS, hereditary elliptocytosis (HE) and hereditary ovalocytosis and membrane transport dysfunction (hereditary stomatocytosis) (Fig. 2) [1620]. Defects that interrupt the vertical structure (spectrin-actin interaction) underlie the biochemical and molecular basis of HS, whereas defects in horizontal interactions (skeletal attachment to membrane proteins) cause HE. All RBC membranopathies share common features, including the loss of surface area, change in morphology, and the resultant tendency for splenic sequestration and extravascular hemolysis resulting in chronic anemia of variable severity [2122]. RBC membranopathies are typically diagnosed by peripheral blood morphology and headed toward confirmation by genetic testing.
Hereditary spherocytosis
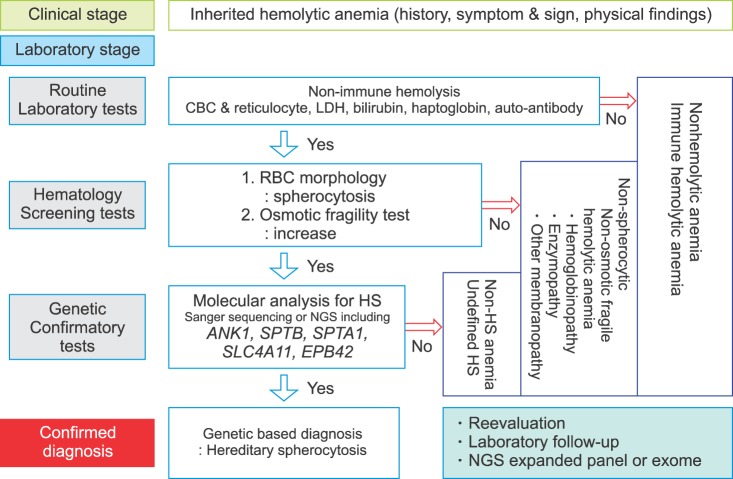
HS is an inherited disorder characterized by the presence of spherical-shaped RBCs on peripheral blood smears and is most commonly associated with autosomal dominant inheritance [23]. HS RBC membranes in HS patients show qualitative and/or quantitative abnormalities of proteins including isolated ankyrin or combined ankyrin and spectrin, isolated spectrin, band 3, and protein 4.2. The defects in the membrane components increase membrane fragility and induce vesiculation with or without the band 3 protein. HS is diagnosed through laboratory tests including RBC morphology and osmotic fragility test as well as family history (Fig. 3). The osmotic fragility test has been considered the gold standard screening test for HS but provides false-negative findings in about 25% of patients [24]. Eosin-5'-maleimide measurement and SDS-polyacrylamide gel electrophoresis of erythrocyte membrane proteins are also useful for screening HS, but standardization of these methods is currently lacking [2526]. None of the HS screening tests can detect all patients because the clinical phenotypes are widely variable, ranging from asymptomatic to severely affected [27]. A few patients with mild hemolysis may develop marked anemia if their bone marrow erythrocyte production is transiently halted by viral (parvovirus B19) or other infections [28]. This scenario would be an aplastic crisis since the bone marrow can no longer compensate for ongoing hemolysis. In neonates or transfused individuals, diagnosis can be difficult due to unclear morphological features and screening tests may be unreliable [23]. Molecular testing is useful for primary differential diagnosis or confirming HS and defining the genotype-phenotype correlation [242930]. The defects in RBC membrane components in HS are typically caused by their corresponding gene mutations. Mutations of the ANK1 (ankyrin 1), SPTB (spectrin, beta, erythrocytic), SPTA1 (spectrin, alpha, erythrocytic 1), SLC4A1 (band 3; solute carrier family 4, member 1), and EPB42 (erythrocyte membrane protein band 4.2) genes have been detected in HS patients [313233]. ANK1 mutations (approximately 50%) are the most common cause of HS, followed by mutations in spectrin genes (SPTB: approximately 20% and SPTA1: approximately 5%), SLC4A1 (approximately 15%), and EPB42 (approximately 10%) [21]. In autosomal dominant HS (75%), nonsense and frameshift mutations of ANK1, SLC4A1, and SPTB predominate. Recessive HS is most often due to compound heterozygosity of defects in ANK1, SPTA1, or EPB42 [34]. Most identified mutations are private, which means no frequent defect is found, and nearly every family has a unique mutation. Genetic-based diagnosis of HS in 25 Korean HS patients revealed the ANK1 gene mutation to be the most common, followed by heterozygous SPTB genes. The genotype-phenotype correlations were clarified after a combined analysis of their cases and a literature review. Anemia was most severe in patients with mutations in the spectrin-binding domain of the ANK1 gene. Splenectomy was more frequently performed in patients with ANK1 mutations (32%) than in those with SPTB mutations [35]. As discussed later in this review, the current availability of advanced genomic surveys, such as NGS, allows one to overcome the limitations of the current diagnostic methods and provide additional information to assess the pathogenicity of identified genetic variants by comprehensive genotype-phenotype analyses.
Hereditary elliptocytosis
HE is a group of disorders characterized by the presence of elliptical-shaped RBCs [36]. HE has a worldwide distribution but is more common in individuals of African and Mediterranean ancestry [3738]. Most patients are asymptomatic; however, a few neonatal presentations can be dramatic, with jaundice, hemolysis, and hydrops fetalis [39]. In Korea, HE is the cause for 1.4% (6/431) of IHA cases [40] and 15 cases have been reported including a genetically identified family with an SPTA1 mutation [41]. HE is inherited in an autosomal dominant fashion and the majority of HE-associated defects occur due to qualitative and quantitative defects in the RBC membrane skeleton proteins, α-spectrin, β-spectrin, or protein 4.1R. Mutations in SPTA1 are the most common, occurring in 65% of HE cases, followed by mutations in SPTB (30%) and EPB41 (5%) [21]. Interestingly, SPTB mutations identified in HE are located on the tetramerization domain but are distributed between the actin-binding domain and spectrin repeats in HS [35]. Hereditary pyropoikilocytosis (HPP) represents a subtype of common HE as evidenced by the coexistence of both HE and HPP in the same family [42]. Patients with severe HE should be considered for splenectomy; however, some degree of hemolysis persists in post-splenectomy HE patients that indicates an incomplete response to splenectomy [8]. Targeted sequencing by NGS is an efficient approach to identify or confirm the diagnosis of HE and HPP, especially in severe, transfusion-dependent cases where the RBC phenotype cannot be evaluated. In addition, causative molecular diagnosis allows identification of genotype-phenotype correlations in theses heterogeneous disorders and may assist in prognosis determination [43].
RBC ENZYMOPATHY
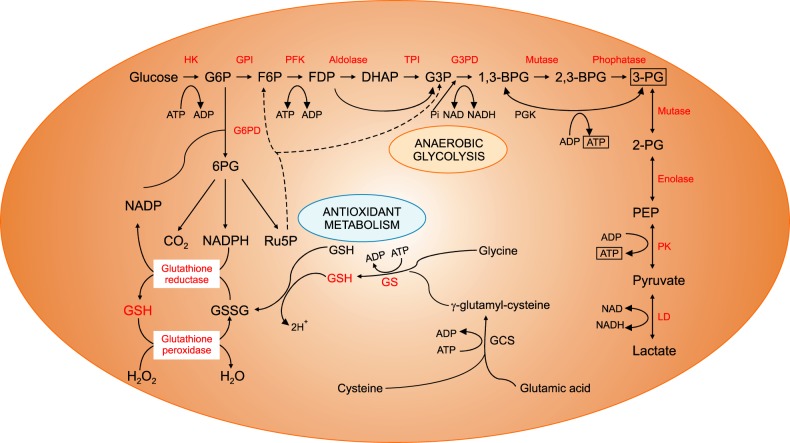
The energy for RBCs is dependent upon the production of adenosine triphosphate (ATP) through glycolysis, and ATP is the only source of energy for the RBCs (Fig. 4). Defects in the glycolytic pathway enzymes have been described in metabolic pathways and almost all are associated with chronic HA [44]. Enzymopathies of the pentose phosphate pathway and glutathione metabolism are associated with acute hemolytic crises after exposure to oxidant substances. Deficiencies or malfunctions of these enzymes generally impair cellular energy balance and/or increase the levels of oxidative stress [45]. Since the discovery of G6PD deficiency in 1956 followed by pyruvate kinase (PK) deficiency in 1961, RBC enzymopathies associated with IHA have been extensively investigated [464748]. The mode of inheritance is autosomal recessive for almost all erythroenzymopathies, except for adenosine deaminase overproduction, which is autosomal dominant and G6PD and phosphoglycerate kinase deficiencies, which are X-linked. In contrast to other IHAs, the morphology of the RBCs shows no specific abnormalities. Diagnosis is based on the detection of reduced specific enzyme activity and molecular characterization of the defect at the DNA level [4950].
G6PD deficiency
G6PD deficiency is the most common RBC enzymopathy, affecting 400 million people worldwide [5152] and is especially prevalent in areas of high malaria infection [53]. G6PD deficiency has an X-linked recessive mode of inheritance and most known mutations in the G6PD gene decrease the enzyme stability. Since these cells do not have the ability to efficiently synthesize glutathione and replenish their enzyme levels, the sulfhydryl groups of hemoglobin are oxidized and damage the RBC membrane as cells age during their 120-day lifespan in circulation. The other mechanism is the decreased enzyme activity, in which the diminished ability of RBCs to withstand stress increases the risk of destruction by hemolysis. Biochemical analysis of G6PD enzyme activity levels is commonly used to screen children with unexplained persistent jaundice [5455]. The analysis may also be used to help establish a diagnosis for people with unexplained episodes of HA, jaundice, or dark urine [56]. The World Health Organization groups G6PD deficiency into five categories, in which G6PD enzyme activity of less than 10% is considered a severe deficiency [57]. Sometimes, patients showing nearly normal G6PD activity can be overlooked because molecular analyses of G6PD are only considered for episodes of hemolytic crisis [58]. Various exogenous stressors such as a medication, fava beans, or infection may trigger hemolysis [59]. At least 186 G6PD mutations have been documented, most of which (85%, 159/186) are single nucleotide substitutions leading to missense variants [60]. In Korea, seven genetically identified G6PD deficiencies have been reported. The mutations were not derived from shared ancestor but have arisen by independent mutational events [61]. With respect to genotype-phenotype associations, the frequencies of Class I mutations found in exon 10 are significantly higher than those in other exons in G6PD deficiency [60]. Mutations in this region, which encodes the binding interface between the subunits, have a highly deleterious effect on enzyme activity by disrupting the quaternary structure and stability of the protein and are the most easily identifiable in the general population [6263]. The application of NGS is useful to diagnose the common, rare and novel variants in G6PD deficiency [64].
Pyruvate kinase deficiency
PK deficiency is a common enzymatic defect of RBCs. Its clinical features are highly variable, ranging from very mild or fully compensated forms to life-threatening neonatal anemia and jaundice necessitating exchange transfusions. PK deficiencies are caused by PKLR mutations and are the most common cause of congenital non-spherocytic HA. Over 200 PKLR mutations have been described [6566]. Up to 70% of PK-deficient alleles carry a missense mutation followed by splicing and stop codon mutations (13% and 5%, respectively; www.lovd.nl/pklr) [50]. Although a genotype-phenotype relationship has not yet been unveiled [67], patients with homozygous null mutations display severe phenotypes including intrauterine growth retardation, severe anemia at birth, and blood transfusion dependence [68]. Splenectomy can reduce the need for transfusion in transfusion-dependent patients, although it does not eliminate hemolysis [44]. Recently, Korean siblings with compound heterozygous null mutations in the PKLR gene have been reported. They initially exhibited congenital dyserythropoietic anemia (CDA)-associated features and Sanger sequencing of the CDA-causing genes was negative. NGS was applied to efficiently identify PKLR mutations as the causal gene in these patients. Although they had a severe phenotype, the patients were eventually cured by hematopoietic stem cell transplantation combined with splenectomy [69].
RBC HEMOGLOBINOPATHY
The hemoglobinopathies are a group of disorders caused by genetic defects that result in the abnormal structure of one of the globin chains of the Hb molecule. With approximately 7% of the worldwide population being carriers, hemoglobinopathies are the most common monogenic diseases and one of the world's major health problems [70]. They fall into two main groups: thalassemia syndromes and structural Hb variants (abnormal hemoglobins). α- and β-thalassemia are the main types of thalassemia; the main structural Hb variants are Hb S, E, and C [71]. α-thalassemia occurs when a gene(s) related to the α-globin protein are deleted or mutated, which occurs most often in persons from Southeast Asia, the Middle East, China, and in those of African descent [72]. β-thalassemia occurs most often in persons of Mediterranean origin and is caused by β-globin gene mutations. To a lesser extent, Chinese, other Asians, and African Americans can also be affected [73]. Some hemoglobinopathies show abnormal RBC morphologies such as target and sickle cells (Fig. 2D, E). Laboratory tests to diagnose hemoglobinopathies are based on the detection of abnormal Hb and include electrophoresis, isoelectric focusing, and high-performance liquid chromatography [7475]. Molecular diagnosis can be made by a variety of techniques, most commonly by Sanger sequencing of the genes coding α- and β-globin and restriction analysis when possible. Over 1,200 genetic alterations that affect the DNA sequence of the human α-like (HBZ, HBA2, HBA1, and HBQ1) and β-like (HBE1, HBG2, HBG1, HBD, and HBB) globin genes are mainly responsible for the observed clinical heterogeneity [76]. Multiplex ligation-dependent probe amplification has become a favored technique to assess for deletions or duplications and can effectively identify different and unknown types of α-globin gene rearrangements to allow the characterization of previously unsolved α-thalassemia genotypes within the α-globin gene region [7778].
Some patients with Hb variants do not show clinical symptoms of HA. They may be detected incidentally by abnormal laboratory findings including falsely low oxygen saturation and glycated hemoglobin (HbA1c)/glucose results [7980]. More than 600 Hb variants have been reported and database records (HbVar, http://globin.bx.psu.edu/hbvar) provide extensive phenotypic descriptions, biochemical and hematological effects, associated pathology, and ethnic occurrence accompanied by mutation frequencies and references [81]. In Korea, 11 Hb variants have been reported [82]. Some have been detected coincidentally while measuring HbA1 levels, including Hb G Coushatta, Hb Queens, Hb Hoshida, and Hb Yamagata [8384]. Genetic analysis can be a more practical application to confirm Hb variants, including novel variants, especially in patients without typical findings. Recent studies demonstrated that NGS could provide a comprehensive assessment of thalassemia screening strategies with its superiority in for both sensitivity and specificity, indicating that NGS is a competitive screening method, especially among populations with a high prevalence of disease [85]. Along with genetic improvement, several gene therapies for the treatment of hemoglobinopathies are currently in clinical trials or under development, including therapies utilizing gene replacement therapy using lentiviruses and the latest gene editing techniques [868788].
NEXT-GENERATION SEQUENCING FOR THE GENETIC DIAGNOSIS OF IHA
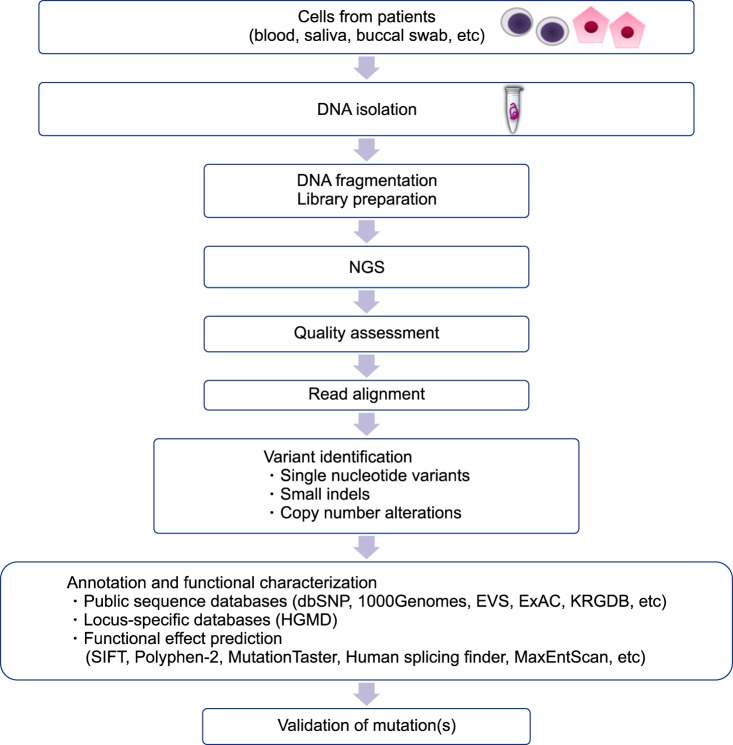
Genetic testing has been used for the confirmatory diagnosis of IHA. Sanger sequencing is primarily performed in order to identify the causative mutations in single gene disorders. It is very lucky to identify mutation(s) in the disease-associated gene in the initial trial. If not, a gene-by-gene approach is required [69]. In these cases, patients may undergo multiple rounds of testing for different genes, a pathway to diagnosis, which can be costly and time-consuming. Additionally, the usefulness of Sanger sequencing is limited for the diagnoses of complex, multi-gene disorders or those with locus heterogeneity. Recent advances in molecular technologies have helped to identify unexpected candidate genes in numerous inherited disorders including IHA [438990]. Various NGS-based methods have been developed, including whole genome sequencing, exome sequencing, and gene panels [91]. Although many recent, successful applications of whole genome sequencing have been reported in establishing the etiology of complex diseases and guiding therapeutic decision-making in neoplastic and nonneoplastic diseases, its use remains challenging and must be carefully evaluated before its clinical implementation as a diagnostic test [92]. Whole-exome sequencing has unveiled numerous causal mutations and genetic modifiers of disease severity in various disorders including IHA [69939495]. Gene panels are currently an attractive option in clinical laboratories because of their reasonable costs, relatively acceptable turn-around time, decreased complexity of data analysis, better coverage over the regions of interest, and reduced incidental findings [969798]. NGS panels consisting of common disease-causing genes have been developed and applied to routine molecular diagnosis for undiagnosed IHA patients and their families [1599]. In particular, patients with the co-presence of membranopathy, enzymopathy, and/or hemoglobinopathy [25100] can be effectively diagnosed using this new technology. Causal gene identification can be deduced through an efficient and reliable strategy to impute and analyze NGS data [101] (Fig. 5). The expanded implementation of the new technology will increase our knowledge of the genetic and genomic differences among individuals, gradually leading to a shift in the clinical management and the therapeutic plan from a population-based approach to a personalized therapy for individual patients [102]. However, it cannot be overemphasized that each laboratory should develop a clinical-grade NGS panel and validate its performance, including analysis and interpretation, before clinical application. A validation strategy is needed to fulfill the requirements set out by the global and national standardized guidelines for NGS panels [103].
CONCLUSIONS
Over the years, IHAs caused by RBC membranopathy, RBC enzymopathy, and RBC hemoglobinopathy were screened and diagnosed by using conventional methods including RBC morphology, membrane protein analysis, Hb electrophoresis, and measurement of RBC enzyme levels. In the genetic era, Sanger sequencing became useful for detecting genetic mutations that cause IHAs. Recent advances in genetic technology utilizing NGS enabled us to better identify various genetic mutations that can cause IHAs. The accurate diagnosis of IHA will be feasible by the use of NGS and associated genetic analyses in the clinical laboratory that could overcome the inconclusive and less accurate morphologic and biochemical analysis. We believe that understanding IHA on genetic basis and applying genetic technologies for routine clinical laboratory testing will improve the accuracy and efficiency of IHA diagnosis and give us insights for precision medicine of each affected individual.
 XML Download
XML Download