

 PDF
PDF ePub
ePub Citation
Citation Print
Print


INTRODUCTION
Autoimmune hemolytic anemia (AIHA) is a rare pathology characterized by an autoimmune-mediated destruction of red blood cells (RBC). The estimated annual incidence of this disease is 1–3 per 100,000 individuals, with a mortality rate of 11% in patients without treatment [1]. According to the characteristic temperature reactivity of the RBC, AIHA is classified into warm AIHA (wAIHA), cold AIHA (cAIHA), and mixed AIHA [2345]. Depending on the underlying cause of the disease, AIHA can be further classified as primary (or idiopathic) or secondary [6].
The wAIHA represents 60% of AIHA cases, and 50% of the patients with secondary wAIHA have lymphoproliferative neoplasia or other autoimmune disorders as the underlying cause [123]. Alternatively, cAIHA is subdivided into primary and secondary cold agglutinin syndrome and paroxysmal cAIHA [5].
AIHA is diagnosed based on certain clinical and laboratorial parameters [57]. Additionally, in order to distinguish between cAIHA and wAIHA, the direct antiglobulin test (DAT) is essential; in wAIHA, the DAT is usually positive for IgG or IgG and C3d, while in cAIHA, the DAT is generally only positive for C3d [578].
Due to the relatively small number of studies on wAIHA, the management of this disease is mainly based on expert opinion [9]. Corticosteroids are considered the first-line treatment for this disease, with 70–80% of patients achieving a complete response (CR). Among the patients with a CR, only 1 out of 3 individuals maintain it, once the steroids are suspended [10]. Second-line therapies should be considered in those patients who require more than 10 mg of prednisone a day to maintain remission and in those that have multiple relapses [10]. Splenectomy and rituximab are the most widely used second-line therapies [111213].
Due to the lack of studies in Hispanic populations with wAIHA and the limited number of publications that have assessed the clinical and laboratorial differences between primary and secondary wAIHA, we decided to conduct this study in order to provide insight into the clinical and biochemical characteristics (i.e., differences between primary and secondary wAIHA) of patients with wAIHA in a tertiary referral center in Mexico City.
MATERIALS AND METHODS
Patients
This was a single-center retrospective study in the Department of Hematology and Oncology at the Instituto Nacional de Ciencias Médicas y Nutrición Salvador Zubirán. We included data for all patients with a definite diagnosis of wAIHA between January 1992 and December 2015. The inclusion criteria were the following: patients aged 16 years or older and diagnosed with wAIHA with a hemoglobin (Hb) level of 11 g/dL or less, features of hemolysis (low haptoglobin level, elevated lactate dehydrogenase [LDH] level, and/or elevated bilirubin level), and a positive DAT with a weak (+/++) or strong (+++/++++) IgG pattern or an IgG+C3d (mixed) pattern.
All the individuals included in this study were evaluated for the presence of systemic lupus erythematosus (SLE) and antiphospholipid syndrome (APLS) based on the following markers: antinuclear antibodies (ANA) (>1:80 UI/mL), anti-cardiolipin antibodies (ACA) IgG (>5.4 UGPL [IgG antiphospholipid units]) and IgM (>4.4 UMPL [IgM antiphospholipid units]), beta-2 glycoprotein 1 antibodies IgG (>2.5 U/mL) and IgM (>3.4 U/mL), and a positivity for lupus anticoagulants. The patients were also evaluated for the presence of the human immunodeficiency virus (HIV), hepatitis C virus (HCV), and hepatitis B virus (HBV). Patients were excluded from the study when incomplete hemolytic profiles or clinical data were encountered during data collection, as well as when other causes of acquired or hereditary hemolytic anemia were determined.
Ethics approval
This study was approved by the institutional review board of ethics and investigation of the Instituto Nacional de Ciencias Médicas y Nutrición Salvador Zubirán.
Treatment
All patients received corticosteroids as the first-line treatment. Methylprednisolone was given in three dosages (1 g/day), followed by prednisone 1 mg/kg/day for 30 days, and the response was evaluated on the third week of treatment based on the Gruppo Italiano Malattie EMatologiche dell'Adulto (GIMEMA) criteria [9]. A CR was considered with a Hb level of >12 g/dL and a normalization of all the hemolysis markers, while a partial response (PR) was considered with a Hb level of >10 g/dL or an increment of 2 g/dL over the basal Hb without the need for transfusions. Based on the above criteria, time to response was defined from the date of the beginning of treatment to the date of response (CR or PR). The absence of response or without response was defined as the lack of complete or partial response. Meanwhile, relapse was defined as a decrease in the Hb level or the appearance of hemolytic markers after the patient achieved a CR or PR. Disease-free survival (DFS) was defined as the time from any response (CR or PR) to relapse [9].
Statistical analysis
The Student's t-test was used for comparing means, and Pearson's coefficient was used for the correlation between groups. The two-tailed Chi-square test and Fisher's exact test were used to compare non-normal categorical data between groups while Spearman's coefficient was used for the correlation between groups. The log-rank test and the Breslow test were used in the analysis of survival parameters. A P≤0.05 was considered statistically significant. The statistical analysis was performed with the SPSS software package, version 21.0 (IBM SPSS Inc., Chicago, IL, USA).
RESULTS
Patients
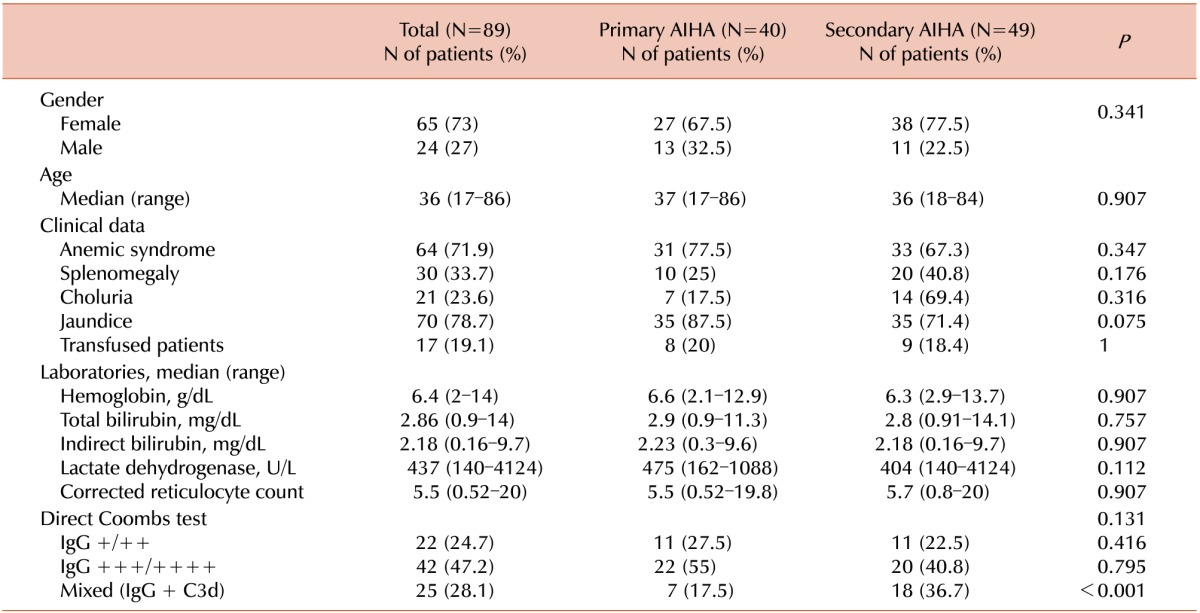
A total of 89 patients were included, 73% of whom were female. The clinical characteristics of the overall population with wAIHA are summarized in Table 1. The median age at diagnosis was 36 years (range, 17 86 yrs). Primary wAIHA represented 44.9% of the cases. Among the 49 patients diagnosed with secondary wAIHA, 32 had SLE or APLS (17 with SLE and 15 with primary APLS) while hemolytic anemia was attributed to other autoimmune disorders in 10 patients (5 with hepatic disease, 3 with thyroid disease, 1 with Sjogren's disease, and 1 with psoriasis) and neoplastic diseases in 4 patients (3 solid malignancies in the gastric, renal, and central nervous systems and 1 hematologic malignancy, namely, non-Hodgkin lymphoma). In the remaining 3 patients, AIHA was attributed to HIV infection, rheumatic fever, and ingestion of allopurinol, respectively.
Biochemical profiles
At the time of diagnosis, the following median parameters were encountered: Hb of 6.4 g/dL (range, 2–14 g/dL), total bilirubin (TB) of 2.86 mg/dL (0.9–14 mg/dL), indirect bilirubin (IB) of 2.18 mg/dL (0.16–9.7 mg/dL), LDH of 437 U/L (140–4,124 U/L), and a reticulocyte index of 5.5% (0.52–20%). When comparing the clinical and biochemical characteristics between the patients that had primary and secondary wAIHA, the only significant difference was the greater frequency of a DAT mixed pattern in secondary wAIHA (Table 1). When comparing the clinical and biochemical characteristics among the different causes of secondary wAIHA, we found that the 10 patients with other autoimmune diseases had higher levels of TB compared with those with SLE/APLS, neoplastic, or other diseases (5.96 mg/dL vs. 2.46, 2.67, and 2.5 mg/dL, respectively; P=0.019) (data not shown). Moreover, we found no statistical differences in survival (data not shown).
Treatment responses
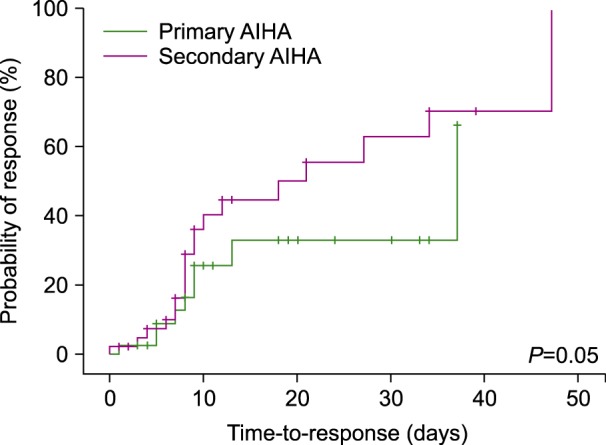
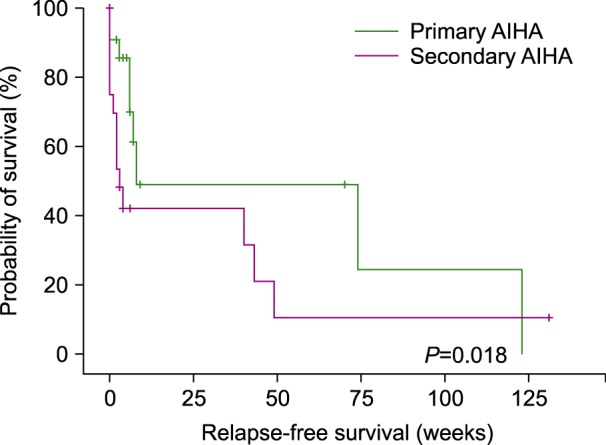
The global response (GR) of the patients receiving the first-line treatment was 84.3% with 50.6% CR and 33.7% PR. The median time to response was 27 days (range, 9–44 days). The patients in the primary group responded in 37 days (CI 95%, 2.92–71.07) whilst the secondary group responded in 18 days (CI 95%, 2.24–33.75) (P=0.05) (Fig. 1). The median DFS for primary and secondary wAIHA was 50.95 weeks (CI 95%, 16.8–85.05) and 28.51 weeks (CI 95%, 7.2–49.82), respectively (P=0.018) (Fig. 2).
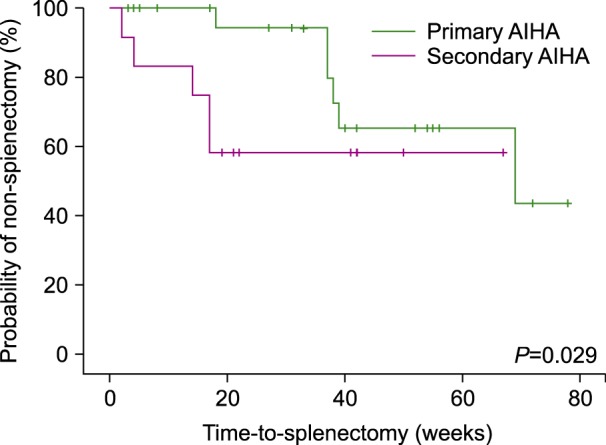
Among a total of 36 patients (40.4%), 12 with secondary wAIHA and 24 with primary wAIHA underwent a splenectomy; 94.4% achieved a GR; 61.1% with a CR; and 33.3% with a PR. The median time from diagnosis to splenectomy was 61 weeks (CI 95%, 50.47–71.54) for primary wAIHA and 43.5 weeks (CI 95%, 27.72–59.43) for secondary wAIHA (P=0.029) (Fig. 3).
Finally, 32 individuals required third line treatment. The third-line treatment consisted of various immune suppressors (i.e., cyclophosphamide, vincristine, azathioprine). Among the patients who received one of the third-line treatment, 93.7% achieved GR, 43.7% a CR and 50% a PR. Due to economic constraints, rituximab was used in only two patients and was categorized as a third-line treatment in our institution.
DISCUSSION
Due to the rarity of AIHA, only few studies have analyzed the epidemiological and biochemical properties as well as the responses to treatment. Furthermore, there are no clinical guidelines that could be useful in the diagnosis, treatment, and evaluation. To date, the largest study was conducted by the GIMEMA group, where 308 patients with hemolytic anemia of various serological types were analyzed. In that study, the most common type was wAIHA [9] and thus we analyzed emphacizingon the wAIHA.
The majority of patients afflicted with wAIHA were female [12131415]. This gender predilection could be attributed to the fact that women are more susceptible to autoimmune diseases. The median age at diagnosis was 36 years, which is slower than the previous reports (i.e., 50 yrs) [1617]. This discrepancy can be explained by the fact that our institution is a referral center for SLE and APLS, which are diseases that mostly affect women of childbearing age.
The most frequent clinical findings in this study were anemia and jaundice. Regarding the patients' hemolytic profiles, median Hb, TB, IB, and LDH were similar to the median values in other reports [718].
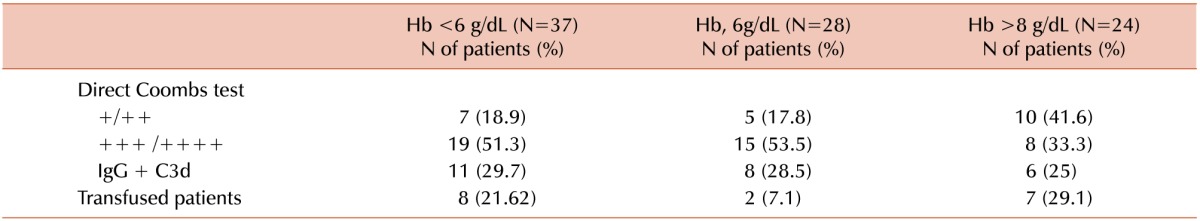
In this study, a positive DAT with a weak pattern was encountered in 24.7% of the cases, a strong pattern in 47.2%, and a mixed one in 28.1%, similar to that of a French study [6]. The presence of a strong pattern correlated with a greater hemolysis (Table 2). Although it was tempting to transfuse these patients based on the levels, we decided for transfusion based on the clinical manifestations caused by the anemia, not solely on the absolute values of Hb, as exemplified in Table 2 and following the recommendations of other centers [69].
Few studies have compared the clinical and biochemical differences between primary and secondary wAIHA [61619]. In our population, the only statistically significant difference encountered between these two groups was the correlation of a mixed pattern with secondary wAIHA (P<0.001). We believe that the greater expression of a mixed pattern in secondary wAIHA was due to the the preexisting autoimmune disease which generated the necessary conditions for a constitutive activation of the complement system through IgG-C3 (and the activator of the classical pathway [C1q]) [20]. Roumier et al. [6] also noted that a mixed pattern was more prevalent in secondary wAIHA.
As stated previously, 84.3% of our patients responded to steroids (50.6% with a CR and 33.7% with a PR). These results are similar to the response rates achieved in other centers, where more than 80% of wAIHA cases responded to steroids [81516]. We also found that patients diagnosed with secondary wAIHA responded faster than those with primary wAIHA (18 vs. 37 days, P=0.05). Nevertheless, probably due to the underlying autoimmune disorders, secondary wAIHA patients relapsed more frequently than primary wAIHA patients that the primary wAIHA patients had a median DFS of 51 weeks versus 28 weeks in patients with secondary wAIHA (P=0.018). It is important to state that other studies have found no statistically significant differences in terms of DFS between primary and secondary wAIHA [1621]. Among the causes of secondary wAIHA, we only found a statistically significant difference in TB levels at diagnosis in the patients with autoimmune-related wAIHA. We believe this could be due to the autoimmune nature of these diseases, or even the concomitant hepatic diseases mentioned previously.
In our center, 40% of the patients were splenectomized, and this procedure represented the standard second-line therapy. The percentage of patients splenectomized by the GIMEMA group was 10%, and they reported a median time from diagnosis to splenectomy of 25 months (92 wks) [8], which does not reflect in time the impact of steroid use; in this analysis, the average time from diagnosis to splenectomy was 15.2 months (60 wks) for primary wAIHA and 10.8 months (43.5 wks) for secondary wAIHA (P=0.029), indicating an unfavorable evolution of secondary wAIHA. Only few studies have evaluated the use of splenectomy as the second-line treatment for AIHA, which makes it difficult to compare our results with published studies due to the heterogeneity of the population and the selection criteria for splenectomy as the second-line treatment [622]. The only study group that evaluated splenectomy was the the Centre de Reference National des Cytopenies Auto-immunes de l'Enfant (CEREVANCE) study, which indicated that the rate of splenectomy in pediatric patients with AIHA is 13.9% [23]. However, they analyzed different AIHA etiologies and we believe that the results of this study cannot be extrapolated to an adult population.
In relation to the third-line treatment in our center, as reported in the literature, there is no standard treatment and the most commonly used are vincristine, cyclophosphamide, azathioprine, and mycophenolate [29]. It can be observed that patients who are subjected to a third-line treatment have a lower percentage of GR and lower percentage of CR.
Despite its retrospective nature, the present study is the first to define the clinical and biological characteristics of wAIHA in a Latin American population and is one of the few that presents the differences between primary and secondary wAIHA. Furthermore, this study established that secondary wAIHA patients relapsed in a lesser time compared with primary wAIHA patients, suggesting that secondary wAIHA patients may benefit from a longer low-dose steroid maintenance period. In secondary wAIHA, treating the underlying disease first is currently recommended, followed by using a monoclonal anti-CD20 antibody (rituximab) as the second-line treatment if a relapse occurs or the disease is refractory.
Despite splenectomy being considered safe and effective in our center, it should be deferred in secondary wAIHA patients because of the associated risks of developing infectious or thrombotic complications. Due to the rarity of AIHA, multicenter prospective studies are necessary in order to achieve a greater number of patients and thus develop better clinical guidelines.
 XML Download
XML Download