

 PDF
PDF ePub
ePub Citation
Citation Print
Print


TO THE EDITOR: Primary cutaneous CD4+ small/medium T-cell lymphoproliferative disorder (CD4+ PCSM-TLPD) is characterized by the proliferation of small-to-medium-sized T-helper lymphocytes within the dermis. According to the 2016 World Health Organization (WHO) classification, the prognosis of CSMTLPD is considered excellent, and it should not be diagnosed as lymphoma [1]. We studied three cases of CD4+ PCSM-TLPD, focusing on its clinicopathological characteristics and differential diagnoses.
Cases
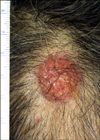
Most patients were middle-aged men who presented with solitary, reddish nodules in the head (Fig. 1). In all three cases, radiological evaluation revealed intradermal cellular nodules, without involvement of the surrounding soft tissue or bone. The nodules of three cases measured 2.7 cm, 1.5 cm, and 0.6 cm in the greatest dimension. In two cases, complete excisions were performed without adjuvant treatment. Both patients were followed-up for 5 and 9 months, respectively, and no adverse events were reported during this period. Case 3 showed complete remission after excision and local radiotherapy, with no evidence of disease for 89 months (Table 1).
All three cases were characterized by a typical dense, nodular, or diffuse lymphoid infiltrate, involving the entire dermis (Fig. 2A). Intraepithelial lymphocytosis was sparse; therefore, no definite epidermotropism was observed. There was a grenz zone just under the epidermis (Fig. 2B). The infiltrate was composed of small-to-medium-sized lymphocytes with mild pleomorphism and was occasionally admixed with B cells and plasma cells (Fig. 2C). The immunohistochemical profile of the tumor cells was as follows: CD20-, CD3+, CD4+, CD8-, PD1+, CXCL13+, and Bcl2+, supportive of follicular helper T-cell (TFH) phenotype. The Ki-67 labeling index was less than 10%. (Fig. 2D-J).
Discussion
The CD4+ PCSM-TLPD was first described in 1995 by Friedmann et al. [2] as a ‘primary cutaneous CD4+ small/medium T-cell lymphoma’, and this entity was classified as a provisional lymphoma in the previous WHO classification [3]. Patients with CD4+ PCSM-TLPD usually present with solitary, reddish tumors, commonly located on the head and neck, with rare instances of ulceration. Spontaneous resolution was observed after an incisional biopsy, and the overall 5-year survival rate was reported to be 60–100%. In particular, the localized lesions showed excellent prognosis, prompting some to consider CD4+ PCSM-TLPD to be a form of reactive lymphoid lesion. With the present knowledge, it remains unclear if CD4+ PCSM-TLPD is a precursor of lymphoma, representing a subtype of cutaneous T-cell lymphoma, or if it is an entirely benign reactive condition (pseudolymphoma) [4].
Garcia-Herrera et al. [5] reported five patients who died of CD4+ PCSM-TLPD. The patients with poor prognosis had characteristic features, such as a larger lesion (>5 cm), higher proliferation indices, decreased expression of CD4, and more monotonous infiltrates, with significantly decreased numbers of background inflammatory cells. The cases we studied did not demonstrate any of the above features.
The differential diagnoses of CD4+ PCSM-TLPD comprise a spectrum of characterizations, including distinct lymphomas, such as the primary cutaneous acral CD8+ T-cell lymphoma, peripheral T-cell lymphoma not otherwise specified (PTCL, NOS), and nodular phase of mycosis fungoides (MF), to benign reactive lymphoid hyperplastic conditions, such as T-cell pseudolymphoma.
Primary cutaneous acral CD8+ T-cell lymphoma may be clinically indistinguishable from CD4+ PCSM-TLPD. However, it presents with a more monotonous infiltrate with a CD8 (cytotoxic) phenotype. Clinically, this type of T-cell lymphoma commonly develops in the ears. In both diseases, the lesions are dermal nodules with no distinct epidermotropism, and show an indolent course [6]. The long-term follow-up of both diseases shows excellent outcomes; complete remission was observed in 100% of the cases (22 CD4+ PCSM-TLPD and 3 CD8+ T-cell lymphoma cases) [7].
Primary cutaneous follicular helper T-cell lymphoma and cutaneous angioimmunoblastic T-cell lymphoma (AITL) share the same immunophenotype (PD1, ICOS, CXCL13, CD10, and Bcl6). Similar to nodal AITL, both diseases show Epstein–Barr virus positivity. Overall, patients with AITL have an aggressive form of the systemic disease [8].
The tumor phase of MF may histopathologically be confused with that of CD4+ PCSM-TLPD. MF lesions are characterized by dense, dermal infiltrates of small-to-medium-sized CD4+ T cells. Despite the histopathological similarity, the clinical features help distinguish these two entities. Patients with MF have a long history of patches and plaques, typical of MF [9]. The histomorphology of the patients studied herein showed nodular proliferation without distinct epidermotropism.
In conclusion, the prognosis of CD4+ PCSM-TLPD is favorable, and it should be differentiated from other aggressive forms of the disease, such as cutaneous T-cell lymphoproliferative disorder and pseudolymphoma. A clinicopathological correlation is very important to avoid diagnostic pitfalls, especially in a small punch biopsy.


 XML Download
XML Download