

 PDF
PDF ePub
ePub Citation
Citation Print
Print


TO THE EDITOR: Primary cardiac amyloidosis accompanying heart failure, angina, and/or arrhythmia is very serious and has a poor prognosis [1]. Sequential heart and autologous stem cell transplantation has resulted in some promising outcomes in a few series [234]. We present a case of primary amyloidosis with cardiac involvement that was successfully managed with these combined approaches.
Case
A 62-year-old woman was referred to our clinic with 3 months of dyspnea on exertion; she was categorized in New York Heart Association class III, and had abnormal echocardiographic findings. She had no other medical history of note. On initial physical examination, her vital signs were as follows: blood pressure, 88/45 mmHg; pulse rate, 79 beats/min; and body temperature, 36.6℃. Neck vein engorgement and pretibial pitting edema were noted. Heart and lung sounds on auscultation were normal. Initial laboratory tests were as follows: white blood cell count, 5,600/µL; hemoglobin, 12.4 g/dL; platelet count, 150,000/µL; protein, 6.0 g/dL; albumin, 3.6 g/dL, blood urea nitrogen, 21 mg/dL; serum creatinine, 1.33 mg/dL; aspartate aminotransferase, 19 IU/L; alanine aminotransferase, 20 IU/L; alkaline phosphatase, 128 IU/L; troponin I, 0.041 ng/mL; and brain natriuretic peptide (BNP), 629 pg/mL. There were no abnormal findings on urinalysis.
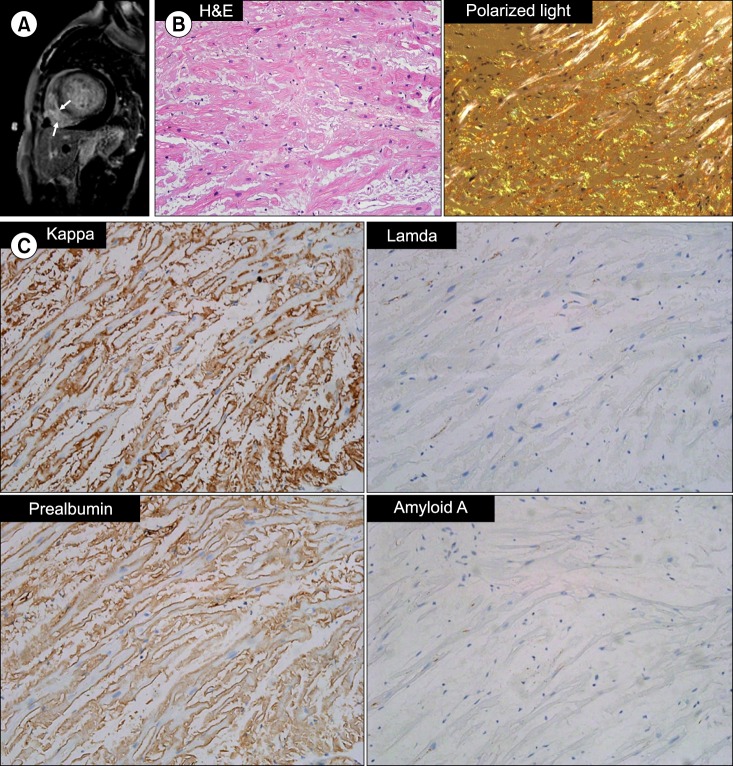
A chest radiograph revealed cardiomegaly with a cardiothoracic ratio of 0.7 and increased interstitial markings suggesting pulmonary edema. Both costophrenic angles were blunted with bilateral pleural effusion. Electrocardiography displayed low voltage in leads I, II, and III and T-wave inversion in leads V5 and V6. Transthoracic echocardiography revealed thickened ventricle walls with minimal pericardial effusion and impaired diastolic function. Left ventricle (LV) filling pressure was high, with an E/E' of 37. No regional wall motion abnormality was observed and the LV ejection fraction was 59%. Cardiac magnetic resonance imaging indicated diffuse transmural or subendocardial enhancement at both ventricular walls on a delayed enhancement image, which were consistent with cardiac amyloidosis (Fig. 1A). Endomyocardial biopsy with a femoral venous approach was performed to confirm this diagnosis. On pathologic examination, amyloid deposits were confirmed by Congo-red staining. Immunohistochemical staining results were as follows: prealbumin (+); kappa chain (++); lambda chain (-); and amyloid A (-) (Fig. 1B, C). Although paraproteinemia or Bence-Jones proteinuria were not evident by electrophoresis and immunofixation, the patient's serum free light-chain ratio was increased to 114 (kappa, 2,040.0 mg/L; lambda, 17.9 mg/L). A bone marrow biopsy revealed mild plasmacytosis of 3.6% without infiltration of neoplastic plasma cells or amyloid deposits. There was no evidence of light chain deposition in other organs including the gastrointestinal tract. The patient had no symptoms suggestive of neuropathy. Computed tomography of her chest, abdomen, and pelvis also revealed no abnormal findings other than benign-appearing hepatic cysts. From these aforementioned findings, the patient was diagnosed with immunoglobulin light chain (AL) amyloidosis with cardiac involvement.
Heart transplantation followed by high-dose chemotherapy (HDC) with autologous stem cell transplantation (ASCT) was the planned treatment in this case. While the patient was waiting for her heart transplant, she received high-dose dexamethasone to control her monoclonal gammopathy, as no other novel anti-myeloma therapies were covered by National Health Insurance in Korea at that time. After 2 cycles of treatment, the patient's intraventricular septal thickness and ejection fraction were unchanged on follow-up echocardiography and her BNP level had actually increased (Fig. 2). However, the difference between the kappa and lambda serum free light chain decreased from 2,022.1 to 8.2 after 4 cycles of high-dose dexamethasone.
Four months after her diagnosis, the patient underwent heart transplantation. Subsequent echocardiography and cardiac computed tomography revealed no abnormal findings. The level of BNP decreased to within normal range. The patient then received immunosuppressive therapy including tacrolimus, mycophenolic acid, and methylprednisolone. Mycophenolic acid was tapered after 8 months. Eleven months after transplantation, the patient underwent high-dose chemotherapy (melphalan 200 mg/m2), supported by ASCT with a well-functioning transplanted heart. She experienced grade 3 diarrhea and grade 3 esophageal ulcers during ASCT, which resolved without sequelae. Neutrophil engraftment (absolute neutrophil count >500/µL) was achieved on day 9. The patient was in complete remission with normal heart function at 21 months after ASCT at the time of last follow-up.
Discussion
Deposits of amyloid fibrils from the monoclonal protein induced-impairment of organ function can cause disease-related morbidity and/or mortality in patients with systemic AL amyloidosis. Cardiac involvement is reported in up to 50% of amyloidosis cases and is associated with poorer clinical outcomes compared with amyloidosis cases involving other organs [5]. HDC with ASCT has been a main therapeutic approach for amyloidosis and has dramatically improved overall survival outcomes from 12–21 months to 4.6 years [678]. However, a substantial degree of treatment-related mortality due to heart failure and/or arrhythmia is associated with this therapy in cases of cardiac amyloidosis, and this has resulted in a much shorter median survival of 1.6 years compared with amyloidosis involving other organs [69]. In addition, advanced stage cardiac amyloidosis is notorious for having a dismal prognosis with a median overall survival of only 5 months [10].
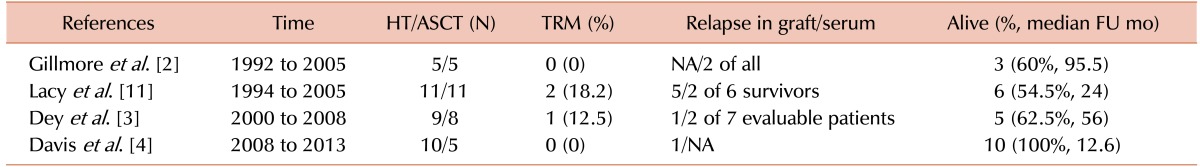
Accordingly, heart transplantation prior to HDC and ASCT has emerged as a treatment strategy for cardiac amyloidosis. In 2006, Gillmore et al. [2] reported that of 5 AL amyloidosis patients with cardiac involvement who underwent heart transplantation prior to ASCT, 3 had long-term survival with no evidence of amyloid deposits. Subsequent promising outcomes have been documented, but mostly from studies in the USA (Table 1) [3411]. To the best of our knowledge, the present study is the first Asian case report of a cardiac amyloidosis patient treated with heart transplantation followed by ASCT. Given the poor prognosis associated with cardiac amyloidosis, this combined treatment modality is likely to be the best available therapeutic option for long-term survival in appropriately selected patients.

 XML Download
XML Download