

 PDF
PDF ePub
ePub Citation
Citation Print
Print


INTRODUCTION
Myeloid sarcoma (MS), also known as granulocytic sarcoma or chloroma, is an extramedullary tumor mass composed of malignant myeloid precursor cells. MS is a rare type of myeloid neoplasm reported in 2.5–9.1% of patients with acute myeloid leukemia (AML). MS generally presents as the first manifestation of AML at the time of diagnosis or at the time of relapse or progression of AML [1]. Less commonly, myeloproliferative neoplasms or myelodysplastic syndrome can transform into MS. Isolated MS is a unique presentation characterized as “non-leukemic MS,” and is defined as the presence of a myeloblastic tumor or infiltration into a single or in multiple extramedullary organs without any evidence of leukemia in peripheral blood (PB) or bone marrow (BM). The period between initial diagnosis of isolated MS and development of AML varies from 0.5 to 24 months (median 3–9 mo) [2].
Although modern histological techniques have improved, difficulty in diagnosing MS has been frequently reported, and 46–75% of MS cases are initially misdiagnosed, most commonly as a malignant lymphoma or even as non-hematopoietic tumors. Because of the limitation of diagnosis by conventional light microscopy, the exact incidence of MS has not yet been determined [3]. Therefore, immunohistochemical analysis is important for lineage definition and differential diagnosis.
Isolated MS can occur at any age of population from pediatric to elderly patients. The most commonly involved sites are the gastrointestinal (GI) tract, skin, bone, soft tissue, and lymph nodes (LNs) [45]. Its clinical presentation can vary depending on the site of involvement. Although various treatment modalities, including systemic chemotherapy, radiotherapy, surgery, or even allogeneic hematopoietic stem cell transplantation (HSCT), have been employed in treating isolated MS, prospective studies to suggest a proper treatment strategy have not been conducted owing to the rarity of the disease. Additionally, the lack of information regarding associated genetic abnormalities deters both clinical management and investigations into molecular mechanisms underlying isolated MS [6]; hence, therapeutic decisions have traditionally relied on retrospective studies, case reports, and small case series [7]. In this study, we described the clinical characteristics and treatment outcomes of 9 patients with isolated MS diagnosed at our institution.
MATERIALS AND METHODS
Patients
A total of 497 patients were diagnosed with AML at Severance Hospital, Yonsei University, from March 1994 to November 2015. Nine of 497 patients (1.8%) with isolated MS who met the following 3 criteria were enrolled: pathologically confirmed extramedullary MS, fewer than 5% BM blasts, and no history of AML. All patients' electronic medical records were reviewed to investigate the clinical and pathological presentations, including age, gender, past history, physical examination, laboratory studies, treatment, and outcomes. MS was diagnosed by conducting immunohistochemical analysis of fixed, paraffin-embedded tissue sections. Immunohistochemical analysis was performed by applying antibodies against myeloperoxidase (MPO), lysozyme, leukocyte common antigen (LCA), terminal-deoxy-nucleotidyltransferase (TdT), CD3, CD4, CD30, CD43, CD56, CD68, CD79a, CD99, and B-cell lymphoma-2 (BCL-2). Cells were considered positive for each marker if the marker was expressed by at least 20% of the neoplastic cells. Following the histologic confirmation of MS, BM biopsy and aspirate with karyotype and gene rearrangement analysis were performed to rule out other hematologic malignancies. Gene rearrangement analyses for RUNX1-RUNX1T1, CBFB-MYH11, and BCR/ABL were conducted using polymerase chain reaction (PCR). Fluorescence in situ hybridization (FISH) and other molecular studies were not routinely performed at the time of histologic diagnosis of MS. Flow cytometric analysis on BM cells was performed at the time of diagnosis of AML.
Complete remission (CR) for MS was defined as complete disappearance of all detectable clinical and radiographic evidence of disease and disappearance of all disease-related symptoms. Partial remission (PR) was defined as a reduction equal or more than 50% of the sum of the products of the greatest diameters of bidimensionally measurable lesions. Overall survival (OS) was measured from diagnosis to death or the last follow-up. Progression-free survival (PFS) was measured from diagnosis until progression to AML.
Imaging analysis
All imaging was performed at the radiology department in our institution. Computed tomography (CT) was performed in 8 patients, magnetic resonance imaging (MRI) in 2 patients, and 18fluorine-labeled glucose positron emission tomography (PET) scanning in 2 of the 9 patients. Three patients underwent imaging with multiple modalities. CT scans were obtained in the axial plane by using a helical CT scanner with 2.5-mm-thick sections after the intravenous administration of iodinated contrast material. MRI was performed with a 1.5-T magnet (Signa Advantage; GE Healthcare, Milwaukee, WI, USA).
Statistical analysis
Statistical analysis was performed using SPSS version 20.0 (IBM Corp., Armonk, NY, USA). Categorical variables between the 2 groups were compared using the 2-sided Fisher exact test, and continuous variables using the Mann–Whitney U test. A P -value <0.05 was defined as statistically significant for all analyses.
RESULTS
The important clinical and laboratory characteristics of all patients at the time of their isolated MS diagnosis are shown in Table 1. The median age was 38 years (range, 24–69 yr); 5 patients (55.6%) were men and 4 (44.4%) women. The primary involved sites of isolated MS could be classified into 4 groups as follows: head and neck (N=4, 44.4%), anterior mediastinum (N=2, 22.2%), gastrointestinal (GI) tract (N=2, 22.2%), and bone (N=1, 11.1%). Cytogenetic analysis of BM cells, which were routinely performed at the time of MS diagnosis, revealed a normal karyotype (NK) in all evaluable patients.
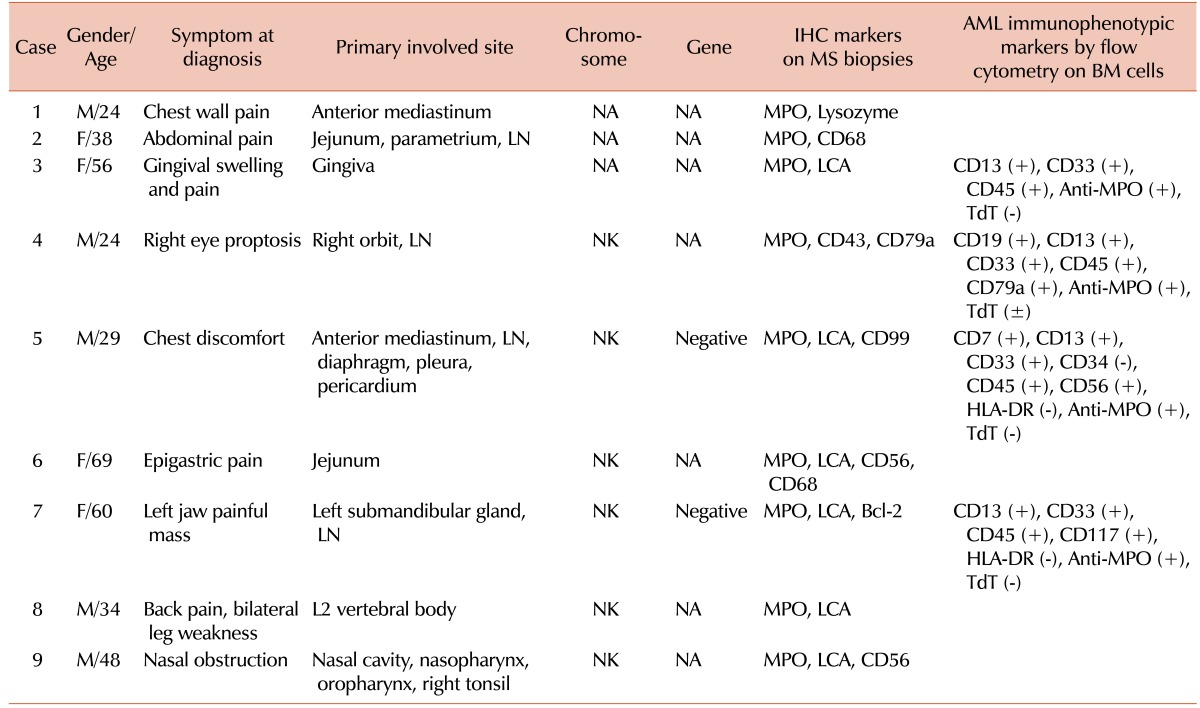
The presenting symptoms, such as chest pain, abdominal pain, gingival swelling and pain, proptosis, painful mass, back pain, weakness in both legs, and progressive nasal obstruction, were mainly related to the primary involved site of disease. Immunohistochemical staining of myeloid origin cells showed variable expression of MPO, lysozyme, LCA, CD43, CD56, CD68, CD79a, CD99, and BCL-2. The immunophenotyping of BM cells revealed myeloid antigen-associated profiles of AML (Table 2).
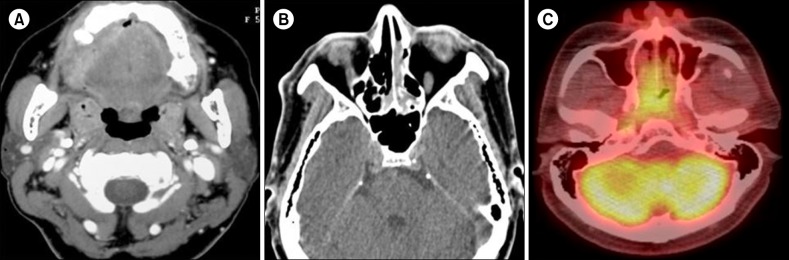
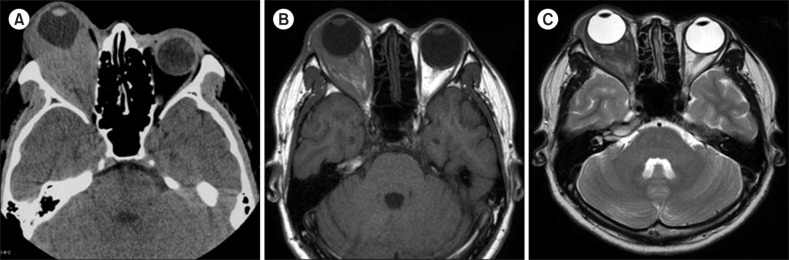
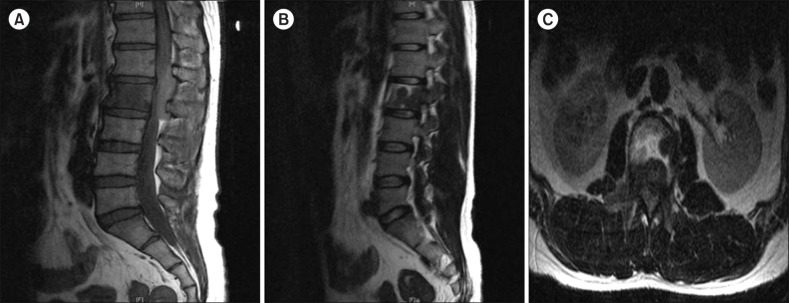
On both CT and MRI, most of the MS lesions (N=7, 77.8%) were shown as discrete solid masses with variable enhancement. Primary gingival involvement was found in one case in which lytic destruction of alveolar bone was also noted on CT (Fig. 1). Isolated MS in the nasal cavity and naso-oropharynx tended to cause diffuse thickening rather than discrete masses on CT. The nasal cavity and naso-oropharyngeal lesions were iso-dense to muscle on CT (Fig. 1). On 18fluorine-labeled glucose PET-CT scans, strong focal uptake was observed in the nasal cavity and naso-oropharynx (Fig. 1). On MRI, both orbital and spinal lesions were documented as well-defined or ill-defined soft tissue masses (Fig. 2, 3). Compared to adjacent muscle, the orbital lesions seen on pre-contrast CT scans were not attenuated by iodinated contrast material. Compared with the periorbital muscle, the lesion also showed an iso-intense signal on T1-weighted imaging and mild hyper-intense signals on the T2-weighted imaging (Fig. 2). A spinal MS in the epidural and paravertebral area was iso-intense and hyper-intense to muscle on T1- and T2-weighted MRI, respectively (Fig. 3).
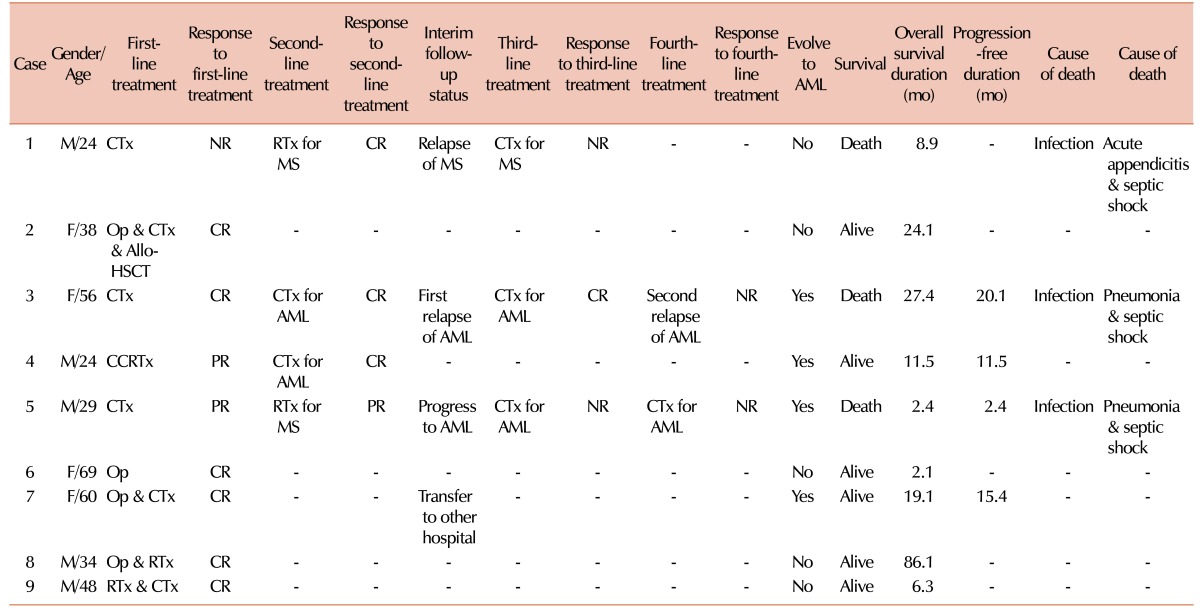
Various first-line treatments to induce remission, such as medical, surgical, radiological, or combined treatment, were administered to patients upon diagnosis of isolated MS. Two patients (22.2%) were treated with local treatment alone, such as surgery and/or radiotherapy, and 7 (77.8%) with systemic treatment. After the first-line treatment, 6 patients (66.7%) achieved a CR, 2 (22.2%) achieved a PR, and 1 (11.1%) failed to respond. The median time from diagnosis to the first CR was 3.6 months (range, 0.2–13.3 mo). The tumors of 4 patients (44.4%) eventually evolved to AML after a median time of 13.4 months (range, 2.4–20.1 mo) from MS diagnosis (Table 3). Two patients who received local treatment alone have maintained their CR status without recurrence of MS (Table 3).
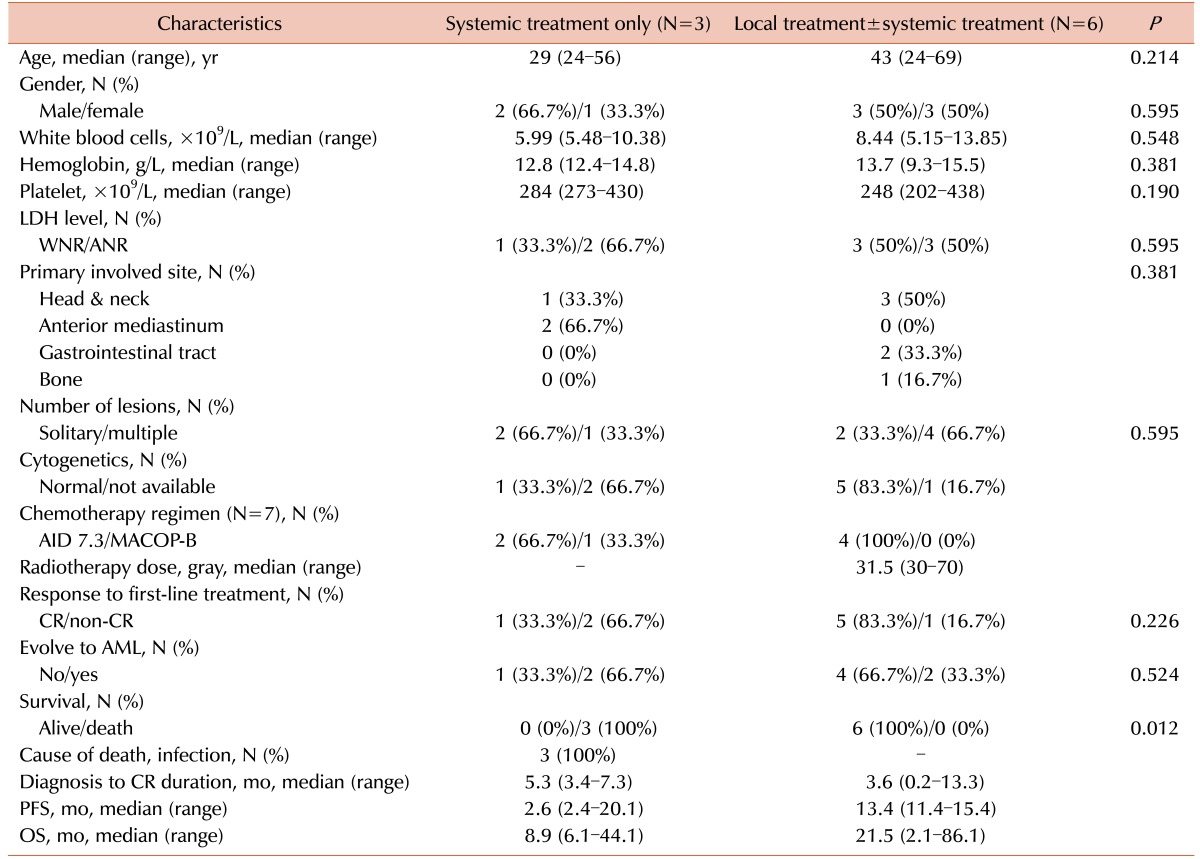
The patients with isolated MS were divided into 2 groups according to the first-line treatment strategy: patients with tumors who received systemic treatment only (S group, N=3) those who received local treatment with or without systemic treatment (LS group, N=6). In the LS group, the median age tended to be greater than that in the S group (43 vs. 29 yr, P =0.214). The male to female ratio was similar between the 2 groups. The number of patients belonging to the head and neck group of the primary involved site was higher in the LS group (N=3, 50%) than in the S group (N=1, 33.3%), although this difference was not significant (P =0.381). There was also no statistical difference in the number of lesions between the 2 groups (P =0.595). After first-line treatment, the number of patients who achieved a CR was higher in the LS group (N=5, 83.3%) than in the S group (N=1, 33.3%) (P =0.226). The number of patients who evolved to AML was higher in the S group (N=2, 66.7%) than in the LS group (N=2, 33.3%) (P =0.524). On follow-up, all patients in the LS group survived, but those in the S group died (P =0.012) (Table 4). Because of the small number of patients in our study, meaningful results could not be obtained on multivariate analysis.
The patients were followed up for 2.1 to 86.1 months, with a median follow-up duration of 21.5 months. The median survival time for all patients was 19.1 months (range, 2.1–86.1 mo) when calculated from the diagnosis of isolated MS, and 3.6 months (range, 0.3–74.4 mo) from the diagnosis of AML (Table 1). The median OS time was longer in the LS group (21.5 mo) than in the S group (8.9 mo). Furthermore, the median PFS time was longer in the LS group (13.4 mo) than in the S group (2.6 mo). Three patients (33.3%) died of infections; however, for these patients, treatment-related mortality was not involved (Table 3).
DISCUSSION
In this study, we have described the overall clinical characteristics of patients with isolated MS treated in our hospital. MS is a rare myeloid neoplasm that is composed of immature cells of the myeloid series and occurs in very different clinical settings. In particular, isolated MS is likely underdiagnosed, and thus, an accurate prevalence rate is difficult to determine. It has been estimated to affect 2 out of 1,000,000 adults and 0.7 out of 1,000,000 children [8]. The age-adjusted incidence rate of isolated MS is 0.9% with a median age of 59 years [9]. The cases described in this study, which displayed MS alone without evidence of AML, account for 1.8% of all AML patients diagnosed in our hospital from 1994 to 2015. Isolated MS has been reported to occur most often in men at the median age of 56 years (range, 1 mo –89 yr) [10]. The male gender predominance (55.6%) of our study population is similar, but our median age of 38 years (range, 24–69 yr) is lower than that of previous studies.
A review of previous studies revealed the high misdiagnosis rate of isolated MS. According to Neiman et al. [11], an accurate initial diagnosis was made in only 27 (44%) of 61 patients. Of the remaining 34 patients, 31 were initially misdiagnosed with malignant lymphoma, 1 with malignant histiocytosis, 1 with synovial sarcoma, and another with Ewing's tumor. Because isolated MS frequently exhibits features mimicking a lymphomatous cell proliferation without definite morphologic evidence of granulocytic differentiation, diagnostic differentiation from malignant lymphoma is very difficult [811]. Because of these limitations of morphological diagnosis, immunohistochemical analysis is necessary during pathological assessment of specimens to differentiate MS from lymphomas or non-hematopoietic tumors. It is also important to determine whether naphthol AS-D chloroacetate esterase staining is negative to show the immature nature of the tumor cells [12].
In this study, immunohistochemical staining for myeloid markers, such as MPO, lysozymes, LCA, and CD68, was performed. Cell surface markers including CD4, CD30, CD34, TdT, and glycophorin A are also useful for diagnosis of MS [1]. Among the various markers, MPO, lysozyme, and CD68 are the most sensitive and essential markers for myeloid differentiation [13]. In our patients, 1 case was initially diagnosed as extranodal NK/T-cell lymphoma, but the final diagnosis was corrected to MS through the addition of immunohistochemical analyses. Therefore, the use of multiple diagnostic modalities, including histochemical and immunoperoxidase stains, conventional and FISH cytogenetics, and flow cytometry, can support the accurate diagnosis of isolated MS.
In general, prior to the confirmative pathologic diagnosis through biopsy, various imaging modalities such as CT, MRI, or PET should be performed for the differential diagnosis, and imaging modalities have to be used to evaluate the extent of the disease after diagnosis of isolated MS. Imaging studies for extramedullary masses may be helpful to differentiate MS, lymphoma, metastasis, extramedullary hematopoiesis, and neurogenic tumors. The characteristic imaging findings of extramedullary masses at various sites and diffuse abnormal BM signal intensity help in the diagnosis of MS in association with leukemia. In previous studies, MS in soft tissues were reported to yield iso-dense or hyper-dense signals relative to brain or muscle on unenhanced CT, hypo-intense or iso-intense signals on T1-weighted MRI, and heterogeneously iso-intense or hyper-intense signals on T2-weighted MRI, and were also known to homogeneously enhance after injection of contrast medium [14151617].
In our study, the imaging characteristics on CT and MRI for MS lesions were fairly consistant. Spinal epidural MS and orbital MS were largely iso-intense on T1-weighted MRI and mildly hyper-intense to muscle on T2-weighted MRI. Other soft tissue MS masses, such as anterior mediastinum, small bowel, and submandibular gland, were largely iso-dense to muscle on CT. Because of its excellent soft tissue contrast, MRI is especially useful for the diagnosis of MS. Additionally, given the poor prognosis, an early and exact diagnosis of MS with MRI evaluation will facilitate appropriate treatment and disease control, preferably before the development of systemic leukemia. Therefore, MS should be considered in the differential diagnosis of new, mildly T2-hyper-intense, homogeneously enhancing soft tissue masses, especially in patients with a history of hematologic disease. However, the clinical usefulness of PET or PET-CT scans in diagnosing or differentially diagnosing MS has to be further validated.
MS can occur in any organ or tissue in the body, and the most common sites reported were bone, LN, and skin [18]. The head and neck area, anterior mediastinum, and small intestine, especially the jejunum, were more frequently involved in our patients, as compared with previous reports. Our study showed that the head and neck area tended to correlate with the evolution to AML more so than other sites (75% vs. 25%), but the number of involved lesions in isolated MS was not significantly related to the eventual evolution to AML. These data suggest that the initially involved site, rather than the number of lesions, might be more important when considering the treatment strategy, and isolated MS occurring in the head and neck area especially must be followed more carefully.
Cytogenetic evaluation is important to classify the disease and to predict the outcome of myeloid neoplasms, especially AML. Similar to AML, cases of MS may harbor chromosomal abnormalities with prognostic significance [19]. Previous studies showed that about half of patients (55%) showed various cytogenetic abnormalities, such as monosomy 7, trisomy 8, trisomy 4, mixed lineage leukemia (MLL) rearrangement, inv(16), monosomy 16, 16q-, 5q-, 20q-, and trisomy 11 [10], with the rest having a normal karyotype. However, Pileri et al. [3] found a normal karyotype in 13 cases (46.4%) and Kaygusuz et al. [4] reported no cytogenetic abnormalities in 6 cases (54.5%). Breccia et al. [8] also reported that most patients (83.3%) showed a normal karyotype and only 2 patients in their study (16.7%) revealed chromosomal abnormalities such as 47, XX, +13 and del(20). In our study, 6 patients (66.7%) who underwent BM cytogenetic evaluation at the time of diagnosis of MS had a normal karyotype, but a normal karyotype in these patients did not appear to be correlated with a favorable outcome. Three patients (33.3%) could not have their karyotype evaluated owing to the failure of metaphasic cell cultivation. Performing cytogenetic examination for MS can be difficult if there is no BM involvement, possibly because of the relatively lower percentage of blasts in the BM. Despite the discrepancies between our data and those from previous literature, isolated MS should always be considered for the possibility of evolving into AML [8]. To validate the clinical significance of cytogenetic abnormalities in the pathogenesis or prognosis of MS, further studies with a larger number of patients are necessary.
Because it is similar to the plasmacytoma in plasma cell disorder, MS has been generally thought of as an antecedent disease entity able to evolve into AML; thus, treatment strategies have been mainly focused on inducing a CR to prevent evolution to AML. In fact, if isolated MS is left untreated, it commonly evolves into AML within 1 year [20]. Unfortunately, the optimal treatment for isolated MS has not yet been clearly defined, in part because of the varied clinical presentations. Previous studies have suggested that the use of AML-like intensive chemotherapy in patients with isolated MS has similar results to those with AML. Neiman et al. [11] reported that 26 (81%) of 32 patients who underwent surgical resection or local radiotherapy progressed to AML within 11 months. On the contrary, 25 (58%) of 42 patients administered chemotherapy remained in the non-leukemic stage for more than 11 months, and 8 (19%) did so for more than 2 years. Byrd et al. [21] reported similar results (mean, 10.5 mo) in patients not receiving chemotherapy. Meis et al. [22] also reported that 4 (25%) of 16 patients who received chemotherapy did not develop AML during a follow-up period of 3.5–16 years after diagnosis of MS. Interestingly, in our study, 2 patients (22.2%) who had undergone only local treatment, such as surgery and/or local radiotherapy, did not evolve to AML (Table 3). The CR durations of these patients were 1.8 months and 83.9 months, respectively. These patients were characterized by solitary lesion involvement in the jejunum and L2 vertebral body at the time of diagnosis of isolated MS, unlike other patients in the LS group (Table 4). In contrast, tumors in 4 patients (44.4%) who received systemic chemotherapy treatment had evolved to AML within a median time of 13.4 months. Local treatment, such as surgery or radiotherapy, might play an important role in controlling primary disease and relieving symptoms without significant toxicity and additional risk of evolution into AML [2324].
Although the role of radiotherapy is less clear, it can be considered as one of the treatment options in isolated MS [18]. Wong et al. [25] and Vassiliou et al. [26] reported similar cases of chemotherapy-resistant, isolated mediastinal MS who achieved a CR following additional radiotherapy. In our study, 1 patient with no response to chemotherapy alone and who underwent radiotherapy had no evidence of disease at follow-up. Tsimberidou et al. [7] reported a review of 20 cases of non-leukemic MS from the MD Anderson Cancer Center in which combination treatment with chemotherapy and radiotherapy resulted in better survival than chemotherapy alone. Although there were a limited number of patients in our study, our results suggest that combined systemic and local treatment for selected patients with isolated MS might be a better treatment strategy to achieve a CR, and to possibly cure the disease, as compared with chemotherapy alone.
More aggressive modalities such as allogeneic HSCT are alternative strategies for isolated MS. In previous studies, the mean disease-free survival time of 12 patients who received allogeneic or autologous HSCT was reported to be 27.3 months (12–48 mo) [272829]. Our patient underwent allogeneic HSCT from a human leukocyte antigen (HLA)-matched sibling donor, in addition to surgery and intensive chemotherapy, and remains disease-free after 18 months. Because the role of HSCT has been rarely evaluated in MS and its efficacy is still unclear, additional case studies are necessary to evaluate the significance of HSCT [227].
In conclusion, isolated MS is a rare and unique entity of myeloid neoplasm and there have been no large prospective randomized studies to determine optimal treatment or prognosis. Survival of patients with MS exceeds that of patients with AML, but remains poor. Therefore, we suggest that initially correct and rapid diagnosis using a multiple modalities approach and early initiation of intensive combined treatment may be the optimal strategy to reduce the risk of isolated MS subsequently evolving into AML. To fully understand the characteristics of isolated MS, a larger number of patients in a multinational study are necessary to identify novel and selected treatments.

 XML Download
XML Download