

 PDF
PDF ePub
ePub Citation
Citation Print
Print


TO THE EDITOR: Budd-Chiari syndrome (BCS) is a rare and potentially life-threatening disorder characterized by hepatic venous outflow obstruction [1]. BCS is associated with thrombogenic conditions such as myeloproliferative neoplasms or inherited deficiencies in protein C, protein S, and antithrombin in at least 75% of patients [2]. However, paroxysmal nocturnal hemoglobinuria (PNH) is another well-recognized cause of BCS [3]. PNH is an acquired disorder of hematopoietic stem cells, characterized by chronic intravascular hemolysis, thromboembolic episodes, and varying degrees of bone marrow failure caused by uncontrolled complement activation [4]. Patients with BCS, in whom no other etiological factor has been identified after a thorough clinical and laboratory investigation, are required to be tested by routine flow cytometry screening for PNH in Western countries [5].
Eculizumab, a humanized monoclonal antibody that blocks the activation of terminal complement C5 components, is currently used in the treatment of PNH. Treatment with eculizumab reduces transfusion requirements, ameliorates anemia, decreases the risk of thrombosis, and improves quality of life by resolving the constitutional symptoms associated with chronic intravascular hemolysis [67]. Long-term treatment with eculizumab in patients with concomitant BCS and PNH has shown a favorable safety profile [8910]. To the best of our knowledge, this is the first report of eculizumab treatment in a patient with BCS and PNH in Korea.
CASE
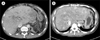
A 39-year-old man was admitted to our hospital with newly developed abdominal pain, fatigue, pancytopenia, abdominal distension, and jaundice. He had a history of liver cirrhosis secondary to BCS and undergone splenectomy and inferior vena cava (IVC) stent insertion 15 years ago. The laboratory results on admission were as follows: white blood cell count, 3.2×109/L; hemoglobin, 5.3 g/dL; platelets, 41×109/L; reticulocyte count, 10.3%; haptoglobin, <100 mg/L (lower limit of reference range, 300 mg/L); lactate dehydrogenase (LDH), 5,005 IU/L (upper limit of reference range, 378 IU/L); and total bilirubin, 55.7 mg/dL (reference range, 0.3–1.2 mg/dL). The findings of an abdominal computed tomography scan and ultrasonography were compatible with BCS, showing underlying liver cirrhosis with the presence of thrombus in IVC stent and hepatic venous congestion (Fig. 1). Despite the continuous treatment of BCS, his clinical condition progressively deteriorated and liver transplantation (LT) was planned. To investigate the cause of the severe hemolytic anemia and pancytopenia, a bone marrow examination and flow cytometric analysis for PNH were performed during preparation for an LT. The bone marrow aspirations showed hypercellular marrow with erythroid hyperplasia and no definite dysplasia. The flow cytometric analysis was performed using antibodies directed against glycosylphosphatidylinositol-anchored membrane proteins (GPI-APs), including CD59, CD24, and fluorescent proaerolysin (FLAER) on granulocytes, and CD59 on erythrocytes. The analysis revealed that 99.4% of granulocytes had FLAER deficiency (Fig. 2), and 5.7% of erythrocytes had a CD59 deficiency. The patient was newly diagnosed with classic PNH and concomitant BCS, and the LT was canceled. He was treated for PNH with intravenous methylprednisolone (62.5 mg/day for 2 days), oral prednisolone (10 mg, 3 times/day), and warfarin (5 mg/day). Following treatment, the patient showed favorable complete blood count results (white blood cells, 3.83×109/L; hemoglobin, 8.4 g/dL; platelets, 120×109/L) and total bilirubin levels (5.1 mg/dL). However, his reticulocyte count (15.6%) and LDH level (5,995 IU/L) were still elevated. Despite steroid therapy, he experienced persistent hemolytic episodes, and eculizumab treatment was initiated. At 3 months of eculizumab treatment, the flow cytometric analysis revealed that 99.9% of granulocytes had a FLAER deficiency, and 50.4% of erythrocytes had a CD59 deficiency. Five months later, improvements in the hemoglobin levels (9.9 g/dL), reticulocytes count (3.0%), and LDH levels (691 IU/L), as well as complete cessation of hemolytic episodes, were observed (Table 1).
This report describes the favorable response to eculizumab in the patient with progressive BCS and PNH. Given the various clinical manifestations and low frequency of PNH, it is inappropriate that every patient with anemia or thrombosis is screened for this disease. However, some clinical presentations, such as BCS or cerebral thrombosis, should warrant PNH testing [11]. According to the retrospective analysis of the South Korean National PNH Registry (N=301), 81 events of thromboembolism occurred in 54 patients (17.9%). Among 81 events, 7 events were identified at the hepatic portal vein (8.6% of events) [12]. Although 424 patients with BCS were identified in Korea during 2009–2013, there is no previous report of PNH with BCS [13]. Thus, routine screening for PNH will be necessary for Korean patients with BCS to examine the exact prevalence of PNH.
Flow cytometric analysis has emerged as the technique of choice for the screening and diagnosis of PNH. At the time of diagnosis, our patient showed 99.4% PNH granulocyte clones and 5.7% PNH erythrocyte clones. The discrepancy of two lineages might result from the complement-mediated destruction of PNH erythrocytes, as compared with other blood cell types resistant to destruction [14]. Additionally, red blood cell transfusions prior to eculizumab treatment might dilute the proportion of PNH erythrocytes, thereby reducing the accuracy of estimating the true proportion of PNH erythrocytes. The monitoring of PNH erythrocyte clones is useful for assessing the efficacy of the response to eculizumab treatment [15]. Since eculizumab inhibits complement-mediated lysis of GPI-AP-deficient cells, more PNH erythrocytes can survive and reflect the true size of PNH clone. In fact, 5.7% PNH erythrocytes at diagnosis had increased to 50.4% in our patient after 3 months of eculizumab treatment. In addition, the patient showed sustained improvements in hemoglobin levels and had not exhibited major episodes of intravascular hemolysis. Given the successful treatment of previous reports [8910] and our case, eculizumab may be tried a primary therapeutic strategy in patients with concomitant BCS and PNH.
In conclusion, to our knowledge, this is the first Korean report of eculizumab treatment in a patient with PNH and BCS. This case supports PNH screening in BCS patients, otherwise the patient could receive unnecessary and aggressive therapeutic options such as liver transplantation. In addition, we demonstrated the utility of eculizumab in the treatment of PNH patients with BCS.


 XML Download
XML Download