

 PDF
PDF ePub
ePub Citation
Citation Print
Print


TO THE EDITOR: Blast crisis may be encountered during the course of chronic myelogenous leukemia, commonly myeloid and less frequently lymphoid or megakaryoblastic. Why or how a patient will progress to myeloid, lymphoid, or megakaryoblastic crisis is still unknown. However, this transition has been attributed to the accumulation of various mutations. The prognosis in such cases varies depending on the type of blast crisis and the molecular signature of the patient. We present a case of CML presenting initially in the chronic phase and progressing to megakaryoblastic crisis in a short period despite receiving treatment.
Chronic myelogenous leukemia (CML) is a clonal disorder of pluripotent hematopoietic stem cells characterized by a balanced, reciprocal translocation involving chromosomes 9 and 22. Most cases present in the chronic phase (CP) of the disease. The progression from CP to the accelerated phase (AP) or blast crisis (BC) may be seen in patients on maintenance therapy due to the accumulation of various mutations. The most common BC type is myeloid, followed by lymphoid, and rarely megakaryoblastic. We report a case of megakaryoblastic crisis in CML, which is extremely rare and has been documented in very few cases to date.
Case
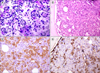
A 42-year-old man presented with complaints of abdominal distension accompanied by fatigue, weakness, and weight loss for the past three months. His medical history did not include any preexisting hematological or medical disorders. Upon examination, he had mild pallor, and a local abdominal examination revealed massive splenomegaly (6 cm below the costal margin). Complete blood counts showed leukocytosis (White blood cells (WBC), 35×109/L), severe thrombocytosis (platelet count, 10×1011/L), and mild anemia (hemoglobin (Hb), 9.5 g/dL). A peripheral blood smear revealed a normocytic red blood cells along with the presence of immature myeloid precursors (differential count: 5% blasts, 17% promyelocytes, 15% myelocytes, 10% metamyelocytes, 2% basophils, 1% eosinophil, and 50% segmented neutrophils). Based on the above findings, a diagnosis of CML was suggested, which was confirmed by PCR for the bcr-abl transcript (Fig. 1A). The patient was orally administered 400 mg of imatinib (Gleevac) and 20 mg/kg of hydroxyurea once daily and was followed up regularly. Upon re-examination after three months of therapy, the patient showed no response to therapy as his cell counts remained unaltered, so he was switched to oral administration of 100 mg of dasatinib once daily. After this, the patient was lost to follow-up and presented after one year with rashes, pain in the upper abdomen and limbs, and general ill health. His counts revealed extreme leukocytosis (WBC: 90×109/L) with anemia and marked thrombocytosis (Hb: 8 g/dL, platelet count: 19.10×1011/L). A peripheral blood smear examination revealed large blasts constituting 65% of total leucocytes along with platelet aggregates, marked platelet anisocytosis, and giant platelets. Bone marrow aspirate was markedly hypercellular with the presence of 80% of blasts having a round nucleus, open-structured nuclear chromatin, prominent nucleoli, and an abundant granular cytoplasm (Fig. 2A). A flow cytometric evaluation was performed on peripheral blood, which identified two populations of blasts. One population (constituting around 40%) was negative for CD45, all B-cell (CD19, CD10, CD20), T-cell (CD4, CD8, CD3, CD7), and myeloid markers (CD13, CD14, CD33, CD117, CD64), as well as CD34, HLA-DR, CD56. The other population of blasts (constituting around 14%) was positive for CD45 (mild) and expressed CD34, HLA-DR, and myeloid antigens (CD13 and CD 33). Subsequently, we also received bone marrow biopsy results, which revealed near complete replacement of hematopoietic precursors with similar blasts with abundant granular cytoplasm, single to multiple vesicular nuclei, and prominent nucleoli (Fig. 2B). Additional immunohistochemical markers, including CD61 and CD68, were also tested. These blasts were strongly positive for CD61 and many of them were also positive for CD34 (Fig. 2C, D). Based on clinical presentation, marrow findings, flow cytometry, and immunohistochemistry, a final diagnosis of CML with megakaryoblastic crisis was confirmed. Additional mutation analysis for imatinib resistance (T315I) by PCR was positive (Fig. 1B). The patient was initiated on induction chemotherapy with adriamycin, etoposide, and cytarabine. However, the patient did not respond well and died of the disease within a month of therapy.
Discussion
In the absence of therapy, most cases of CML evolve to the blast phase. The most common BC type is myeloid, which is seen in 70% of patients, followed by lymphoid or mixed-lineage phenotype [1]. Megakaryoblastic crisis is rarely encountered in these patients and accounts for less than 3% of cases [2]. To date, although various studies have been performed, no specific mutation has been found that could predict the type of crisis in these patients. As compared to myeloid or lymphoid crisis, megakaryoblastic crisis has a poorer prognosis [3]. Hence, an early detection is imperative for timely administration of chemotherapy with the goal of reverting the disease to chronic phase and proceeding to allogeneic stem cell transplantation (SCT) if possible.
The closest differential of CML in BC is the Philadelphia chromosome (Ph+) AML, as both may share common clinical features and bone marrow morphology. Since their management varies widely, various authors suggest caution in diagnosing CML in BC versus de novo Ph+ AML. Clinically massive splenomegaly, thrombocytosis, and basophilia support a diagnosis of CML in BC, as opposed to de novo AML. Although classically described in CML, Ph+ can also be seen in some cases of AML. However, its transcript size in AML is usually 190 Kd, compared to 210 Kd in most cases of CML. The presence of additional genetic alterations associated with the progression to the blast phase may also aid in establishing the correct diagnosis.
The exact mechanism of progression to BC is still unknown. Various studies speculate that stem cells undergo additional genetic alterations leading to transformation to BC. In a study done by Calabretta and Perrotti [4], they noted that the ectopic expression of the p210 bcr-abl transcript resulted in growth factor independence and reduced susceptibility to apoptosis, leading to leukemic transformation of immortal hematopoietic stem cells. This abnormal transcript activated the Ras-MAP kinase and PI3Akt pathway leading to uncontrolled proliferation of leukemic cells [4]. The chromosomal abnormalities that have been found to be associated with progression to the blast phase include duplication of Ph+, trisomy 8 or 19, isochromosome 17q (leading to loss of the p53 gene on 17p), or translocations and inversions associated with MDS/AML [5]. The appearance of these mutations during treatment plays a significant role in the advancement of the disease. In addition, these genetic alterations lead to imatinib resistance in these patients [5]. Not all patients will benefit from TKIs and many patients become resistant to it while on treatment, especially those who show a suboptimal response. A single specific point mutation, known as T315I kinase, in the bcr-abl gene has been isolated in association with resistance to imatinib. Patients who are resistant to imatinib appear to benefit from SCT. Fruehauf et al. [6] observed a more promising hematological response for patients with CML in BC receiving a combination of mitoxantrone with etoposide.
In conclusion, CML in megakaryocytic BC is rarely encountered in clinical practice. It is important to correctly diagnose these cases as they benefit from a combination of conventional chemotherapy with imatinib, resulting in decreased mortality.

 XML Download
XML Download