

 PDF
PDF ePub
ePub Citation
Citation Print
Print


INTRODUCTION
The fusion of the 3´ region of the proto-oncogene c-ABL on chromosome 9q34 with the 5´ region of the BCR (breakpoint cluster region) gene on chromosome 22q11.1 creates Philadelphia chromosome [1], an acquired genetic defect in a hematopoietic stem cell (HSC). It was the first described chromosomal abnormality to be associated with a specific human disease of malignant origin such as chronic myeloid leukemia (CML) [23]. A malignant clonal disorder of pluripotent hematopoietic stem cells causes an increase in circulating cells of myeloid, erythroid, and megakaryocyte lineage in peripheral blood and myeloid hyperplasia in bone marrow (BM) [4].
BCR-ABL1 oncoprotein is a constitutively active tyrosine kinase that perturbs numerous signal transduction pathways resulting in uncontrolled cell proliferation, reduced apoptosis and impaired cell adhesion [5]. Breakpoints in BCR occur mainly in one of the three regions: the major breakpoint cluster region (M-bcr), minor breakpoint cluster region (m-bcr), and micro-breakpoint cluster region (µ-bcr) while breakpoints in ABL occur in the first and second introns. The corresponding fusion mRNA joins exon a2 from the ABL gene to either exon b2 or b3 of BCR gene when BCR is broken at M-bcr (called b2a2 or b3a2 transcripts), or to exon e1of BCR gene (called e1a2 transcript), or to exon e19 of BCR gene (called e19a2 transcript) when BCR is broken at m-bcr and µ-bcr, respectively. Different fusion mRNAs translate into chimeric proteins of different sizes: both b2a2 and b3a2 produce 210 kDa, e1a2 produces190 kDa, and e19a2 produces 230 kDa proteins. The b3a2 and b2a2 account for the vast majority of CML cases [6].
The majority of CML patients (95%) display M-BCR breakpoints resulting in p210 fusion proteins while the remaining (5%) have m-BCR breakpoints corresponding to the p190 fusion proteins [7].
The sole standard therapeutic regimen administers tyrosine kinase inhibitors (TKIs) that specifically target this oncoprotein and inhibit its aberrantly increased tyrosine kinase activity [8]. Thus, an accurate and reliable detection of the BCR-ABL1 fusion protein is highly demanded for diagnosis and monitoring [9].
In the past decade, many new technologies have been developed to replace labor-intensive cytogenetic analysis. These include techniques detecting the BCR-ABL1 fusion at the DNA level with genomic polymerase chain reaction (PCR), Southern blot analysis, fluorescence in situ hybridization (FISH), or chromosomal painting; at the RNA level, Northern blot analysis or reverse transcriptase polymerase chain reaction (RT-PCR); and at the protein level, Western blot analysis or immunoprecipitation [10].
Routinely performed techniques like karyotyping, FISH or quantitative real time PCR (RQ-PCR) are laborious, time consuming and they also require well trained staff. Moreover, these techniques do not detect the aberration at the protein level [5111213].
A new flow cytometric immunobead assay has recently been developed for detection of BCR-ABL1 fusion proteins. This assay detects the fusion proteins in lysates of leukemic cells by use of a bead-bound catching antibody against one side of the fusion protein and a fluorochrome-conjugated detection antibody [141516].
In the present study, we have used this method to detect the BCR-ABL fusion protein on TKI naive and TKI treated chronic, accelerating and blast Phase of CML patients in order to investigate performance of this method in the diagnosis of CML.
MATERIALS AND METHODS
Selection of subjects
During the study period from February 2009 to January 2014, a total of 648 CML (male: 362; female: 286) patients with median age of 38±1 (range 8–78 yrs) were investigated for the status of BCR-ABL1 protein. Among 648 patients, randomly chosen 83 patients were enrolled for the comparative study of BCR-ABL1 positivity by three routinely used procedures including karyotyping, RQ-PCR as well as immunobead flow cytometry assay. Further, 112 TKI naive and 536 follow up patients were included in the study. Follow-up patients were TKI treated for 6–9 months, 12–18 months and 24 months or more. In case of 83 patients of comparative study, 15 were newly diagnosed (TKI naive) and 68 patients were follow up patients, among whom 28 were followed up for 6–9 months, 28 were for 12–18 months and 12 were for 24 months follow up after TKI treatment. The study was approved by the institutional review board and written consent was obtained from each individual.
Sample collection
Three to 5 mL of peripheral blood and/or 2 to 3 mL of BM samples were obtained in EDTA tube from the patients for routine cell morphology analysis, flow cytometry analysis and RQ-PCR. 2 to 3 mL of blood sample was collected into heparin tubes for conventional cytogenetic analysis.
Flow cytometric immunobead assay for detection of BCR-ABL status
Initially, we have counted leukocytes of fresh peripheral blood and/or fresh BM sample (collected in EDTA tubes) and the sample containing 20×106 leukocytes was incubated with lysing buffer (20 times the sample by volume) for 10 min. Then 250 µL of aliquot sample was pre-treated in ice for 10 min with protease inhibitor to maintain the integrity of BCR-ABL1 fusion protein. Thereafter, samples were incubated for 15 min with 100 µL of the 1x stock of the BD lysate treatment reagent (BD Biosciences) and then centrifuge at 20,000 g for 10 min at 4℃. Then, anti-BCR antibody (BD Bioscience) coupled to a bead and Phyco-erithrin (PE) labelled anti-ABL antibody (BD Bioscience) were incubated with the processed samples to recognize the BCR-ABL fusion protein molecules with the help of a FACS Calibur (BD Bioscience). Anti-BCR antibody, anti-ABL1 antibody and prepared sample were mixed in 1:1:1 ratio (50 µL each) and incubated for 2 hours in dark. Then washing was done by 1 mL wash buffer. After that, sample was ready for flow cytometric analysis. Cellquest Pro software (BD Bioscience) was used for this study. The prepared bead immunoassay samples were run on a dual-laser flow cytometer (FACS Calibur) which is capable of detecting forward and side light scatter and fluorescence emissions at both 578 nm and 660 nm and the mean fluorescence intensity (MFI), which expresses the status of BCR-ABL1 fusion protein in the studied samples. Lysates prepared from 140 randomly selected age and gender matched normal healthy blood donors' peripheral leukocytes (WBC) and also BCR-ABL1 negative HL60 cell line were analysed as negative controls. The median fluorescence (PE MFI) was measured. A suspension of the BCR-ABL1 positive cell line (K562) was made and the cell count was done. Blood sample was taken from a normal individual and divided into 3 tubes. By simple mathematical calculation the volume of the cell line suspension was added to the normal blood samples in such a manner that the samples contain BCR-ABL1 positive cells in 1%, 10% and 50%, respectively. The bead immunoassay with these three samples was done by flow cytometry. The mean MFI values were 6.64, 17.94, and 88.36, respectively. Also, during standardization 20 BCR-ABL1 positive peripheral blood samples (confirmed by RQ-PCR) were analysed by bead immunoassay which also showed positive result.
RQ-PCR for BCR-ABL1 fusion gene detection
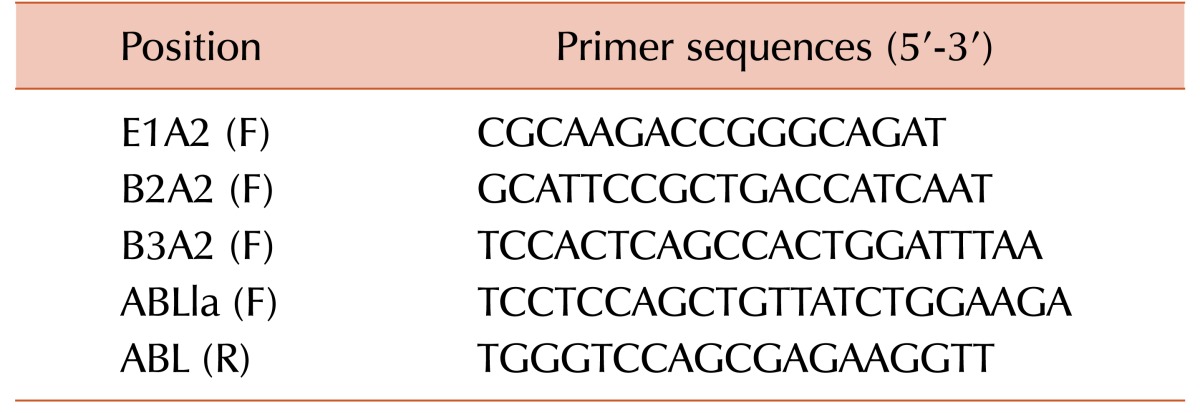
RNAs from randomly selected 83 samples were isolated using Qiagen RNeasy mini kit (Qiagen, Valencia, CA). cDNA was prepared using cDNA synthesis kit (Thermo Fisher Scientific Inc., Rockford, IL, USA), following manufacturer's protocol and RQ-PCR was done for detection and quantification of BCR-ABL1 protein expression with specific break points. SYBR Green (ABS) was used as a dye and hotstar taq (Qiagen) was used as taq polymerase in RQ-PCR. Reaction mixtures of 25 µL contained 12.5 µL of SYBR green, 5 mM MgCl2, 200 µM dATP, dCTP, dGTP, 100 ng template DNA and 300 nM forward and reverse primers. Primers have been listed in Table 1 [17]. The PCR cycler conditions were 98℃ for 15 min, and 45 cycles of 95℃ for 15 sec, 65℃ for 60 sec, 72℃ for 30 sec and a combined annealing and extension step of 72℃ for 5 min.
The RQ-PCR result is expressed as the fold increase of the BCR-ABL1 gene per fold increase of the control gene, ABL [13]. The percentage value of BCR-ABL1/ABL1 ratio is mentioned for interpretation of the results.
The RQ-PCR was performed using BCR-ABL positive cell line (K562) suspension in 1%, 10% and 50%, respectively. The BCR-ABL/ABL (B3A2 transcript) values were 5.64%, 30.26%, and 38.86%.
Conventional karyotyping
Cytogenetic studies were performed on metaphase cells derived from peripheral blood cultures after arresting metaphases by colchicine treatment. Metaphase cells were prepared for G-banding, and karyotypes were interpreted according to the 1995 International System for Human Cytogenetic Nomenclature using Ikaros metasystem software (Carl Zeiss, Oberkochen, Germany). Karyotyping was performed from BCR-ABL positive cells in 1%, 10% and 50% dilutions. The Ph chromosome positive values were 0.61%, 6.95% and 35.8 %, respectively. BCR-ABL negative HL60 cell line was used as negative control in flow cytometry, RQ–PCR, and Karyotyping.
RESULTS
The MFI values of Ph-negative normal controls as well as the Ph negative cell line were obtained and the slightest fluorescence reflected from the normal uncoated beads was also measured. The mean value was 1.3 and SD was 0.032. Thus, 1.36 (mean+2SD) has been accepted as negative cut off value (maximum value of fluorescence: 1.36 and minimum value of fluorescence: 1.24) for this study in our laboratory set-up. Thus, MFI values of Ph-negative normal controls were within the range of 1.0–1.356.
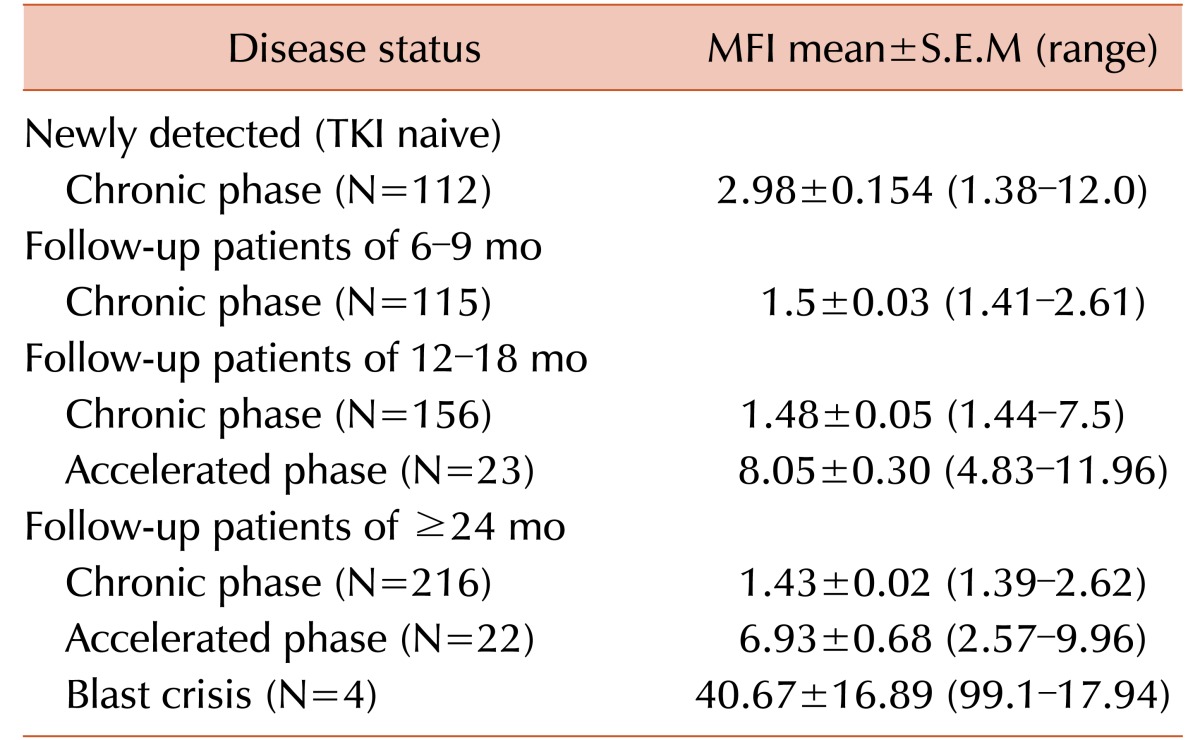
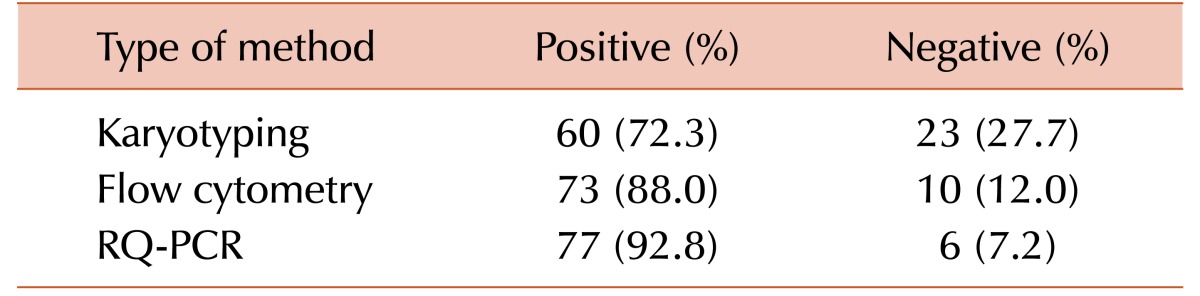
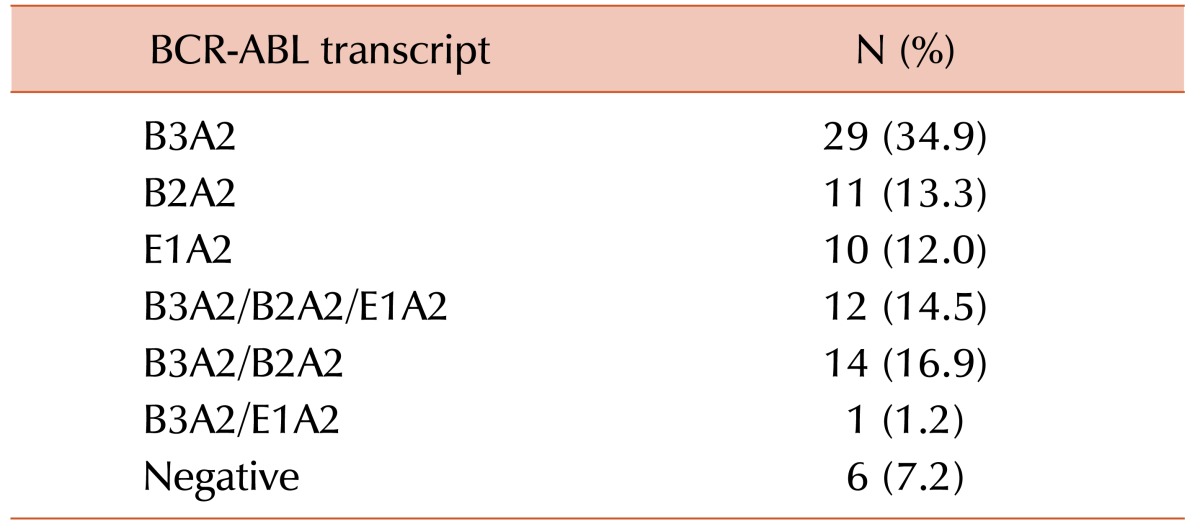
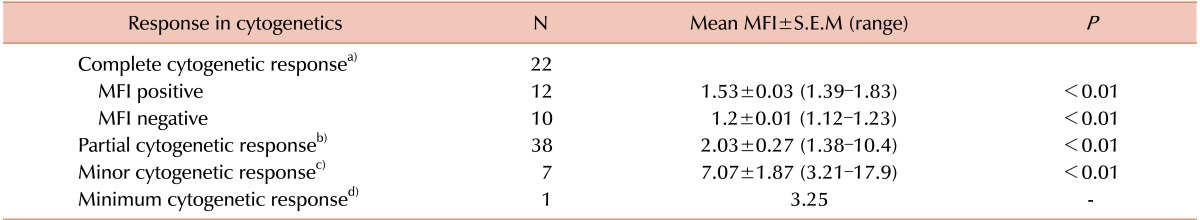
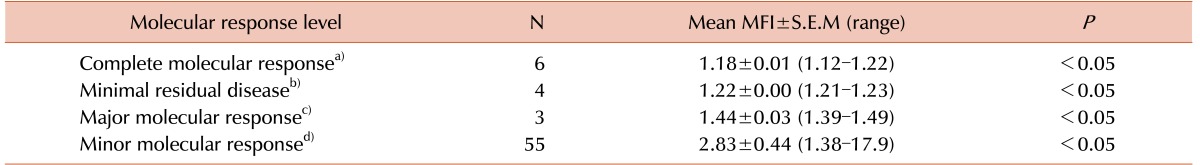
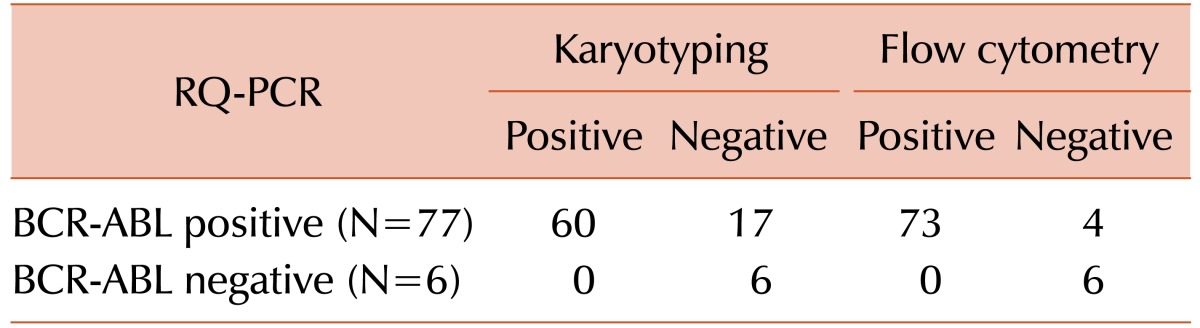
All patients had undergone fluorescence activated cell sorting assay to detect the status of BCR-ABL fusion protein at the time of diagnosis and different times of their treatment period. The mean MFI values and the maximum and minimum ranges of each group are shown in Table 2. The PE MFI of BM samples showed higher threshold value than peripheral blood (PB: 3.34; BM: 3.83), though there was no statistically significant difference between mean PE MFI values of peripheral blood and BM sample (P>0.05). These 83 patients underwent the detection by conventional cytogenetics, RQ-PCR for BCR-ABL1 fusion gene and fluorescence based flow cytometry for BCR-ABL1 chimeric protein and the results are documented in Table 3. Following the ELN guidelines [18], patients were characterized at cytogenetic as well as molecular level. Transcript percentages, detected by RQ-PCR, were depicted in Table 4.
Mean MFI of 15 patients of TKI naive was 3.03±0.29 (mean±S.E.M) and range was 1.62–4.8. According to the disease status, 6 patients were in complete response, 68 were in chronic phase (CP) (including TKI naive patients), 5 patients were in acute phase (AP), one patient was in blast crisis (BC) and 3 patients were in relapse. When we considered the MFI values of different phase groups of these CML patients, we found the mean MFI (±S.E.M) of CP group was 1.75±0.12 (excluding TKI naive CP patients; 1.38–4.8); mean MFI (±S.E.M) of AP group was 7.79±3.40 (3.21–17.9); MFI of BC patient was 6.44 and the mean MFI (±S.E.M) of relapse patients was 5.04±1.18 (3.25–7.26). A significant difference in MFI values is very obvious between patients in CP to other groups including AP, BC and relapse (Table 5, 6).
Executing the cytogenetic response as well as the molecular response of the TKI treated patients, we have analysed the MFI values, ranges and statistical significance levels of different response groups (Table 5, 6) and MFI values showed a strong t correlation with BCR-ABL1 RQ-PCR and cytogenetic test (Spearman's rho=0.847 and 0.786, respectively; significance level or P=0.00).
Below is the ROC curve for peripheral blood sample which represents that the sensitivity and specificity of BCR-ABL1 protein assay is 97% and 100%, respectively. The Area under curve of the ROC was 0.971 (CI: 95%; LB-UB 0.93–1.00). The PE MFI value of 1.36 was considered the best value to discriminate positive and negative samples for peripheral blood using the BCR-ABL1 immune assay.
Consequently, the specificity and sensitivity of the BCR-ABL fusion protein assay have been evidenced (Table 7) showing 100% and 95% using flow cytometry technique respectively whereas conventional cytogenetic study showed 78% sensitivity.
DISCUSSION
Recent CML studies revealed the efficacy of TKI therapy as the choice of first line treatment. The TKI treatment requires, fast and easy way to detect the BCR-ABL1 aberration.However, the BCR-ABL1 aberration is so far being detected by karyotyping, FISH or RQ-PCR; all these techniques are relatively time consuming and require facilities, mainly present in specialized laboratories and consequently requir referral of the samples to diagnostic reference centres.
In the present study we have used a simple flow cytometric immunobead assay for detection of the BCR-ABL1 fusion protein in cell lysates and evaluated by comparing it to RQ-PCR as well as the conventional cytogenetic analysis. Consequently, the efficacy, sensitivity and quantification potentials of the BCR-ABL1 fusion protein assay have been suggested.
The TKI naive and treated patients as well as levels of molecular/cytogenetic response to TKI therapy in the present work have been amalysed and such affirmative findings had never been achieved by the previous researchers who studied only the TKI naive groups and excluded the follow up patients [141920].
MFI values solely depend on the presence of BCR-ABL1 fusion protein, therefore more advanced CML phase showed the higher MFI value, as shown in Table 2. Comparing the MFI values among TKI naive and different TKI treated follow up patients, the mean MFI of TKI naive group was 3.03 whereas TKI follow up patients showed 1.75 in CP patients (excluding TKI naive group), 7.79 in AP patients and 5.04 in relapse patients. The significant difference in MFI values is clearly shown in different groups of disease phases (P<0.000). Patients having highly positive value may grow some kinase domain mutations causing resistance to prescribed TKI and have evolved into accelerated (AP) or relapse phases (data not shown).
The patients who had achieved the complete cytogenetic response (CCYR) within first 6 months and currently still in CCYR and surviving were also analyzed. Those patients who had not achieved the cytogenetic remission within first 6 months did not show good prognosis. Some of them achieved complete response, but some of them were in AP phase. The BCR-ABL1 fusion protein assay showed considerable consistency with RQ-PCR and cytogenetic results. Studying molecular response groups (Table 6), we have found a distinct difference in MFI values such as complete molecular response (CMR) patients are also negative to be detected for BCR-ABL protein by fluorescence based flow cytometry assay (mean MFI 1.24). Mean MFI of mMR group, being 2.61, indicates clearly the difference in response levels.
The discrepancy was that the present BCR-ABL fusion protein assay could not detect the minimal residual disease (MRD) conditions. four MRD were detected negative in MFI status. The transcript levels in MRD patients were too low (0.01–0.005%) indicating the BCR-ABL protein assay could identify only up to 3 log level (0.1%) of the transcripts. The concordance rate of the present technique is 95.18 % to RQ-PCR.
Cytogenetic analysis (Table 5) showed that most of the complete cytogenetic response (CCR) patients were also negative for MFI values as was expected, and PCR, mCR and minCR patients were having high MFI values (mean MFI 2.03, 7.07 and 3.25, respectively) accordingly. But it is well known that the conventional cytogenetic method is much less specific when compared to RQ-PCR technique. Our results of flow cytometry are more consistent with RQ-PCR results as 12 CCR patients and 1 patients of TKI naive onset of disease out of 83 were positive in RQ-PCR and BCR-ABL1 protein assay by flow cytometry but they were Ph chromosome negative by karyotyping. Not only that, one newly diagnosed TKI naive patient was negative for Ph chromosome in cytogenetic analysis but he had positive MFI value (1.67) as well as B3A2 and B2A2 transcripts by RQ-PCR. Thus, the BCR-ABL1 protein analysis technique was found consistent and much more sensitive (16 % greater sensitivity) and reliable than the conventional cytogenetics.
We found no difference in morphologic features, cytochemistry, or immunophenotype between the blasts in PB and BM samples in any of the cases studied. For cases with adequate metaphases, there was strong correlation between the cytogenetic results for PB and BM samples. But peripheral blood can be used as a substitute for a BM aspirate only when BM cannot be obtained and circulating blasts are present in peripheral blood. Because normally 0–5% blasts are present in BM and no blast is found in normal peripheral blood, unless the number of blasts is increased in the BM and they are released into the peripheral blood, conventional karyotyping may give either negative result (in case of major molecular response or minimal residual disease) or less percentage of Ph positive cells.
The present assay can document the effective transaction of the molecular transcript. It allows rapid identification of the presence of the BCR-ABL1 protein (p190, p210, p190/210) [21]. The anti-BCR antibody is developed against a non-homologous region of B80 amino acids, encoded by exon 1 of the BCR gene to detect all known BCR-ABL variants, including p190, p210 and p230 and other rare variants. On the basis of the position of the BCR and ABL epitopes that are recognized by the applied antibodies, it can be anticipated that all rare and novel BCR-ABL variants will be detected by the immunobead assay, as was proven for the detection of novel e18-a2 BCR-ABL (p225) variant [2223]. This is in contrast to the currently used RT-PCR technique, which is focused on the m-bcr (p190) and M-bcr (p210) breakpoints and might easily miss the p225 and p230 variants. Furthermore, the immunobead assay seemed to be specific and sufficiently sensitive to detect BCR–ABL proteins (p190 and p210) in precursor-B-ALL cell lysates and CML [5].
It is promising that the same immune bead assay can be used for detection of other fusion proteins, if the appropriate antibodies can be raised [24]. As differentially labelled beads allow multiplexing, it will be possible to develop single tube assays for combined evaluation of multiple different fusion proteins, for example, per disease category. Consequently, the flow cytometric immune bead assay can contribute to fast and easy diagnosis and classification of leukemias [25].
Another significant observation of our study is that the group of patients with co-expression of two or three transcripts showed comparatively high MFI in immune bead assay. In view of this, we can conclude more than one type of transcripts may be seen in samples with high MFI value. However, the present study proved that the flow cytometric immunobead assay ensures recognition of all variants of the BCR-ABL1 fusion protein independent of their break point position but as we did not find any patient with very rare breakpoints producing the rare fusion proteins such as p195, p200 or p225 in the present study, it is still to be proved in the future.
Our present study has some limitations also; as our study is a retrospective study and some key statistics cannot be measured, and significant biases may affect the selection of controls. Moreover, as we have used SYBR Green dye which may bind to any amplified dsDNA, primer dimers and unspecific products may be detected as fluorescent signal, together with the specific product of interest. Therefore, it is extremely important to have well-designed primers that do not amplify non-target sequences, and that melt curve analysis be performed.
Our present flow cytometric bead immunoassay is proved as an accurate, sensitive, and reliable technique. The ROC curve analysis (AUC is 0.971) indicated the present assay as excellent method with high sensitivity (97%) and specificity (100%). The comparative data of sensitivity (Table 7) shows that our present technique is much more sensitive (about 95%) than the conventional cytogenetic method (about 78%) when we considered RQ-PCR as the gold standard.
In summary, the flow cytometric bead immunoassay can be used in any hospital or laboratory and easy to perform. The assay is independent of the breakpoint position in the BCR gene, does not need special molecular laboratory facilities other than a routine flow cytometer. It provides results within 4 hours, and can be run in parallel with routine immunophenotyping. This is particularly important in countries like India, where molecular diagnostics is not readily available for supporting patient care. As flow cytometry showed about 95% concordance rate to RQ-PCR, in case of flow cytometric detection of minimal residual disease, some sort of lack of reliability is noticed. In the present study 4 MRD patients escaped the detection of BCR-ABL fusion protein by present flow cytometric method. Otherwise, in case of first diagnosis and monitoring the follow up cases, flow cytometry can be run reliably and safely which is easier, quicker and simpler, and fully translatable to routine management of CML patients [26].
In conclusion, the flow cytometric immunobead assay for BCR-ABL1 protein detection may be preferred for investigation of peripheral blood and BM samples of CML patients as it was concordant with RQ-PCR and/or cytogenetic analysis. So, the semi-quantitative BCR-ABL1 fusion protein assay might be used to diagnose and monitor CML patients reliably and safely though further development is essential to achieve a quantitative assay to identify all the MRD cases at the molecular level.
 XML Download
XML Download