

 PDF
PDF ePub
ePub Citation
Citation Print
Print


TO THE EDITOR: Hemophagocytic lymphohistiocytosis (HLH) is characterized by severe immune activation and deregulation resulting in extreme and often life-threatening inflammation [1]. Adult-onset HLH is rare and fatal. Infectious agents contribute to a major part of the etiology of adult-onset HLH [23]. A high degree of clinical suspicion and prompt treatment is required to prevent mortality. We have described an unusual clinical presentation of a case of cytomegalovirus (CMV)-associated HLH with multi-organ involvement.
CASE
A 71-year-old man presented with altered mental status, fever, and multi-organ dysfunction. His symptoms began 2 months ago with gradually progressive fatigue, generalized maculopapular rash, arthralgias, fever, anemia, pneumonia, and acute renal insufficiency at a hospital admitted previously. Preliminary laboratory tests for infection and autoimmune diseases were all negative. The rash and arthralgia developed after a recent travel to India (how long did he stay there?). The physical examination was unremarkable except for left hemiparesis and right-sided Bell's palsy. The initial laboratory results were as follows: hemoglobin, 94 g/L; white blood cell (WBC) count, 15.1×109/L; platelets, 42×109/L; and neutrophils, 90%. The results of his cerebrospinal fluid (CSF) studies were suggestive of meningitis with high protein level, low glucose level, and increased number of lymphocytoid cells. Bronchoalveolar lavage (BAL) fluid culture revealed growth of Klebsiella pneumoniae. After admission, the patient was treated with broad-spectrum antibiotics, steroids, and anti-tuberculosis therapy. Over the next few days, his condition worsened due to anemia, pancytopenia, and abnormal renal and liver functions. His ferritin level was progressively increasing, with a peak level of >3,000 ng/mL. Brain magnetic resonance imaging (MRI) studies revealed sulci expansion and abnormal signals over the subarachnoid spaces. The patient did not have any evidence suggestive of an immunocompromised state.
Considering the progressive pancytopenia, multiorgan dysfunction, low fibrinogen level, and high ferritin level, a provisional diagnosis of HLH was made. His serum soluble IL-2R level elevated to 1,054 pg/mL. Peripheral blood smear revealed normocytic hypochromic anemia with moderate anisopoikilocytosis. Numerous histiocytes with evidence of hemophagocytosis were observed in bone marrow aspiraion without any significant dysplasia (Fig. 1A, B). Granuloma or fibrosis was not observed. Fungal and acid-fast staining were negative. Meanwhile, owing to the worsening pneumonia, the patient underwent follow-up BAL and showed cytomegalovirus (CMV)-positive by polymerase chain reaction (PCR). At the same time, peripheral blood PCR showed 154,111 copies/mL of CMV, and a repeat CSF analysis also showed CMV-positive by PCR. Immunohistochemical staining in the BM biopsy was positive for CMV (Fig. 1C). Overall, the findings were compatible with a diagnosis of disseminated CMV-associated HLH. The patient was admitted to the intensive care unit. However, in spite of all the measures taken, including immunosuppressive treatment with etoposide, and broad-spectrum antibiotics and ganciclovir for CMV, he died 2 weeks after the confirmation of the diagnosis of CMV-associated HLH.
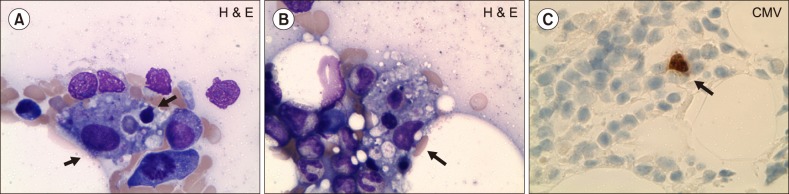 | Fig. 1(A–C) Features of the bone marrow (BM) aspirate and biopsy showing hemophagocytic lymphohistiocytosis (HLH) and cytomegalovirus (CMV) staining. (A, B) BM aspirate showing histiocytic hyperplasia and prominent hemophagocytosis by activated histiocytes. Arrowheads indicate features suggestive of ongoing endocytosis (Wright-Giemsa stain, ×1,000). (C) BM core biopsy result showing positive immunohistochemical stain for CMV showing a large cell with intranuclear inclusions suggestive of CMV in the BM.
|
Go to : 
DISCUSSION
HLH occurs as either a primary (familial) [4] or a secondary (sporadic) disorder [56]. Both conditions manifest pathological immune activation and may be difficult to differentiate from each other. Primary HLH is an autosomal recessive disease with an incidence of 1 per 50,000 live-born children [5]. Younger patients often have a clear familial inheritance or genetic mutation. The median survival is <2 months if untreated. Immunological triggers such as vaccinations and viral infections may trigger bouts of disease in these patients. However, in many circumstances, no clear-cut immunological trigger is identifiable. Secondary HLH [7] includes adults and older children who lack a family history or known genetic cause of HLH. The diagnostic criteria for HLH were mainly derived from studies in the pediatric population, but characteristics of adult HLH are now recognized [8]. Secondary HLH often occurs as a result of pathological immune activation in response to a trigger. The frequently noted triggers include malignancy [9] (especially hematological malignancies, including acute leukemia, myelodysplastic syndrome, and myelofibrosis), infections (especially viruses such as EBV and CMV), and rheumatological disorders [2810]. Immune-activated and immune-mediated pathologies likely play a central role in the evolution of HLH. These represent acute clinical signs and symptoms of immune activation, including hepatomegaly, jaundice, adenopathy, rash, seizures, and focal neurologicneurological deficits, as well as abnormaly high serum level of cytokines such as interferon gamma (IFNγ), tumor necrosis factor α (TNF-α), interleukin 6 (IL-6), IL-10, and macrophage-colony stimulating factor (M-CSF) [1112]. Biopsies of lymphoid tissues or histological examination of liver tissue from HLH patients reveals highly activated macrophages and lymphocytes, supporting striking activation of the immune system [10]. Therefore, the goal of initial therapy is to suppress the hyperactive immune system for preventing immune-mediated irreversible organ damage [13]. Induction therapy is often followed by allogeneic stem cell transplantation if a suitable donor is available. If no suitable donor is identified, patients should be followed up closely for signs of relapse. The HLH-94 protocol proposed in 1997 [14] included an 8-week regimen of etoposide, dexamethasone, and intrathecal methotrexate. The clinical profile of our patient was complex, and HLH in an elderly patient without any preexisting factor supporting an immunocompromised state is unusual. The clinical features that raised the suspicion of HLH were fever, cytopenia, organomegaly, coagulopathy, liver function abnormalities, elevated ferritin level, and hemophagocytic lymphohistiocytic in the BM. Disseminated CMV associated with HLH is uncommon in adults, and anecdotal pediatric cases were reported [15]. Early recognition and treatment of HLH are essential, and rare pathogens such as CMV should be considered as a cause of HLH.
Go to :
 XML Download
XML Download