

 PDF
PDF ePub
ePub Citation
Citation Print
Print


INTRODUCTION
Cancer has become the most common cause of mortality in the twenty-first century. The survival rate of cancer patients has been increasing dramatically along with advances in the early detection and treatment of cancer. Cancer survivors, however, encounter a number of health problems, including the development of a second cancer [1]. Therapy-related myeloid neoplasms (t-MNs) are well-recognized among these secondary malignancies. T-MNs are a distinct disease entity in the acute myeloid leukemia (AML) classification and occur as a late complication of previous cytotoxic treatment for a primary malignant or non-malignant disease [2]. Although t-MNs are subdivided into therapy-related myelodysplastic syndrome (t-MDS), acute myeloid leukemia (t-AML), and myelodysplastic/myeloproliferative neoplasm (t-MDS/MPN) based on disease parameters [23], the diagnosis of t-MN is usually made based on the same criteria used for the diagnosis of their de novo counterparts, with a 20% blast threshold for a diagnosis of AML [4]. The latency period between the primary disease diagnosis and t-MN onset ranges from several months to years, and is associated with the cumulative dose, dose intensity, and type of preceding cytotoxic therapy [5]. Most patients with a t-MN have clonal chromosomal abnormalities in their bone marrow cells, which often correlate with the type of preceding cytotoxic agents and latency period. The t-MNs occurring after exposure to alkylating agents or radiation therapy, known as classical t-MNs, typically present after a latency period of 5 to 7 years and are often preceded by a myelodysplastic phase [6]. On the other hand, other distinct types of t-MN may occur after exposure to topoisomerase II inhibitors, which include both intercalating agents (e.g. doxorubicin and mitoxantrone) and non-intercalating epipodophyllotoxins (e.g. etoposide and teniposide). These t-MNs typically have a shorter latency period of 1 to 3 years with an initial presentation as overt leukemia, often without a preceding myelodysplastic phase [56]. Exposure to topoisomerase II inhibitors is predominantly associated with translocations of the MLL gene on chromosome band 11q23 [4]. In general, patients with t-MNs have shorter survival than that of patients with de novo AML [5]. t-MNs are relatively resistant to conventional therapies and show poor clinical outcomes, with a reported median survival of 8-10 months [37].
As t-MNs are less frequently diagnosed in younger patients, the data regarding t-MNs in the pediatric population are limited [8910]. In this study, we retrospectively reviewed the medical records of patients diagnosed with a t-MN before the age of 21 years. This study aimed to characterize t-MNs arising from childhood cancers and to analyze the outcomes of t-MNs occurring in patients less than 21 years old, an increasingly significant problem encountered by pediatric hematologists.
MATERIALS AND METHODS
Patient characteristics and Diagnosis of t-MN
A retrospective chart review was performed on 16 patients who were below the age of 21 years when diagnosed with a t-MN at Samsung Medical Center between 2002 and 2011. We analyzed patient characteristics, latency period, cytogenetics, and treatment outcomes. All patients were previously exposed to chemotherapy, radiation therapy, or a combination of both. Most patients had been treated for their primary disease at our institution, with the exception of 3 patients who were referred to our institution after having been diagnosed with t-AML at another facility. Treatments administered to patients with t-MNs included myeloid-type induction chemotherapy and allogeneic hematopoietic stem cell transplantation (HSCT), based on the general health and disease status of the patient. The diagnosis of t-MN was made after reviewing the slides of the bone marrow aspirate smears and biopsy sections, immunophenotypic data, and pathology reports. Cytogenetic studies were performed using standard techniques, and karyotypes were described according to the International System for Human Cytogenetic Nomenclature [11]. The t-MN diagnosis was made according to the WHO 2008 classification system; the term t-AML is used when the blast count is ≥20% in either the peripheral blood or bone marrow, with a history of prior exposure to chemotherapy or radiation therapy. The diagnosis of t-MDS was made when the patient had one or more cytopenias and at least ≥10% dysplasia in one or more lineage of peripheral blood and bone marrow with a percentage of bone marrow blasts of <20% [2].
Statistical methods
Latency period was defined as the interval from the first diagnosis of the primary disease to the diagnosis of the t-MN. The length of survival from the date of diagnosis of the t-MN (the first bone marrow examination) to the last follow-up date was also evaluated. Univariate analyses of event-free survival (EFS) and overall survival (OS) were calculated using the Kaplan-Meier method, and comparisons between survival curves were performed using the log-rank test. P-values <0.05 were considered statistically significant.
RESULTS
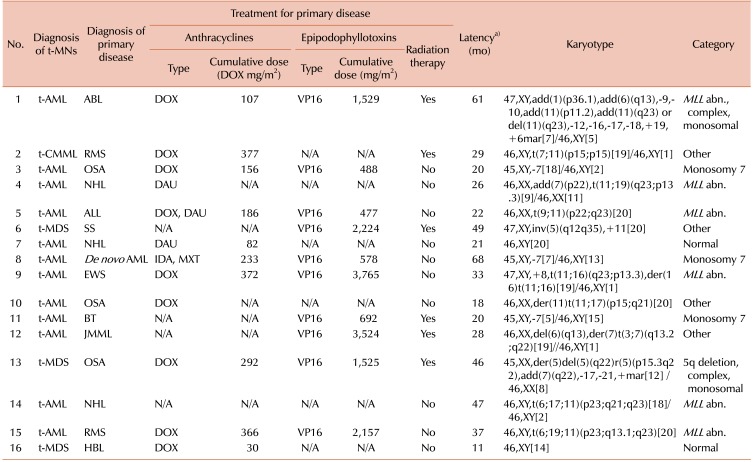
Patient characteristics are summarized in Table 1. The median age at the time of the diagnosis of the primary malignancy was 8.7 years (range, 0.8–18.8 yr), while the median age at the time of the diagnosis of the t-MN was 11.5 years (range, 1.6–20.4 yr). The t-MNs diagnoses were as follows: t-AML in 12 (75%) patients, t-MDS in 3 (19%) patients, and therapy-related chronic myelomonocytic leukemia (t-CMML) in 1 (6%) patient. The primary malignancies were osteosarcoma (N=3, 19%), acute non-myeloid leukemia (N=3, 19%), non-Hodgkin's lymphoma (N=2, 13%), rhabdomyosarcoma (N=2, 13%), brain tumor (N=1, 6%), Ewing sarcoma (N=1, 6%), hepatoblastoma (N=1, 6%), synovial sarcoma (N=1, 6%), juvenile myelomonocytic leukemia (N=1, 6%), and de novo AML (N=1, 6%). Except for 2 (13%) patients who had normal karyotypes, the majority (88%) of cases had cytogenetic aberrations, as shown in Table 2. An 11q23 abnormality was the most frequently observed cytogenic abnormality (N=6, 38%), followed by monosomy 7 (N=3, 19%). One patient (case 8) whose primary diagnosis was de novo AML showed a change in karyotype. Two patients had a normal karyotype (N=2, 13%), and 1 patient had a 5q deletion (N=1, 6%). Primary therapeutic exposure histories are listed in Table 2. For 3 patients who were transferred to our institution after the diagnosis of t-MN, the exact cumulative dose and dosing schedule were not available. Fifteen of the 16 patients had received alkylating agents. All patients had received topoisomerase II inhibitors, either epipodophyllotoxins or anthracyclines, or both. The cumulative doses of anthracyclines converted to doxorubicin isotoxic equivalents ranged from 30 to 377 mg/m2, lower than the usual safe maximum dose of 500 mg/m2. The cumulative doses of the epipodophyllotoxin etoposide in all applicable cases ranged from 477 to 3,765 mg/m2. In addition, 5 patients had concurrently received radiation therapy, and 2 patients (case 1 and case 12) underwent allogeneic HSCT during their primary treatment. The median latency period was 2.4 years (range, 0.9–5.6 yr).
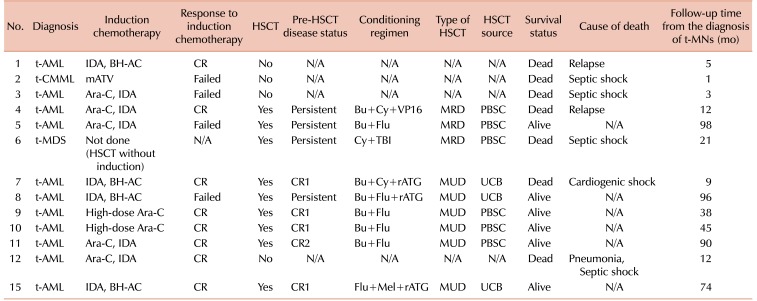
Of the total 16 patients, 13 patients (82%) were treated with curative intent. A detailed description of the treatment for t-MN in these 13 patients is summarized in Table 3. Twelve patients received AML-type induction therapy, while the remaining patient (case 6) underwent allogeneic HSCT without induction therapy. The types of induction therapy included a cytarabine plus idarubucin (IDA) regimen for 5 patients, IDA plus N4-behenoyl-1-beta-D-arabinofuranosylcytosine (BH-AC) for 4 patients, high-dose cytarabine for 2 patients, and a modified ATV regimen for the remaining t-CMML patient. Of the 12 patients who received AML-type induction therapy, 8 (67%) patients achieved complete remission (CR). Meanwhile, 1 patient (case 2) died during induction therapy and 3 patients (cases 3, 5, and 8) failed to achieve CR.
Except for 4 patients who could not proceed to transplantation either due to treatment-related mortality (N=3; cases 2, 3, and 12) or parental refusal (N=1; case 1), 9 patients underwent allogeneic HSCT. As some patients who had achieved CR relapsed before HSCT, 4 patients were in first complete remission (CR1) at the time of transplantation, and the remaining 5 patients were in second complete remission (CR2) (N=1) or in a refractory or persistent (N=4) disease state. Conditioning regimens are listed in Table 3. The most commonly used conditioning regimen was busulfan plus fludarabine with or without rabbit antithymocyte globulin. Among the 9 patients who underwent allogeneic HSCT, 3 had matched-related donors, 3 had matched-unrelated donors, and the remaining 3 patients underwent umbilical cord blood transplantation.
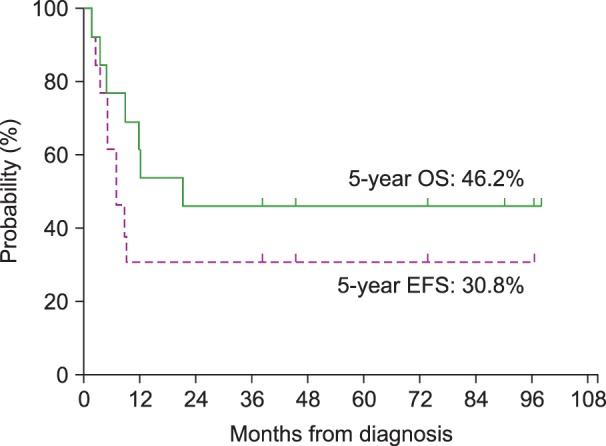
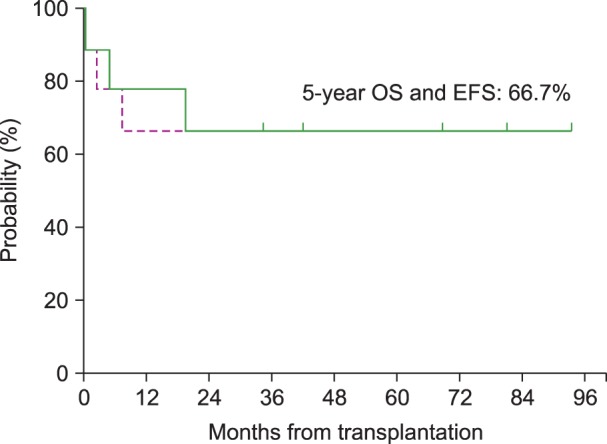
For the 13 patients who received treatment with a curative intent, the 5-year OS and the 5-year EFS were 46.2% and 30.8%, respectively, with a median follow-up of 66 months (range, 24–165 mo) from the time of the diagnosis of the primary malignancy and 21 months (range, 1-98 mo) from the time of the diagnosis of t-MN (Fig. 1). The survival of the 9 patients who underwent allogeneic HSCT was separately analyzed; both the 5-year EFS and the 5-year OS were 66.7% (Fig. 2). In addition, among the 6 patients who received an alternative donor graft (matched-unrelated donor in 3 patients and umbilical cord blood in 3 patients), 5 patients survived. The median follow-up duration after transplantation was 75 months (range, 42–93 mo) among transplant survivors.
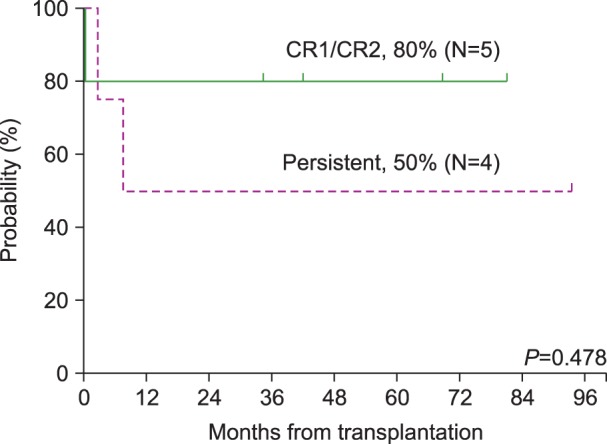
Fig. 3 shows the survival after allogeneic HSCT in terms of disease status at the time of transplantation. Of the 9 patients who underwent allogeneic HSCT, 5 patients were in CR1 or CR2, and 4 patients were in persistent disease status. Among the 5 patients in CR1 or CR2 at the time of transplantation, 1 patient died, resulting in an OS of 80% in this group. This patient died of cardiogenic shock presumably caused by cyclophosphamide-induced cardiomyopathy. For the remaining 4 patients in a persistent disease status, 2 patients died resulting in an OS of 50% in this group. One patient died of septic shock, and the other patient died of leukemia relapse and progression.
DISCUSSION
The t-MNs, a serious long-term complication of previous cytotoxic therapy, have been a widely investigated topic in adults. With the increasing number of pediatric cancer survivors, this disease entity is also an increasing concern in children and adolescents. Pediatric t-MNs are estimated to account for about 1% of all childhood cancers [12], while Hodgkin's disease accounts for over 4% of childhood cancers [13]. This analysis of 16 patients with t-MN represents the first reported single-center series of t-MNs in the pediatric population in Korea.
The primary diagnosis in all 16 patients was malignant disease. While hematologic malignancies comprise nearly 40% of childhood cancer [14], our results did not show a predilection for hematologic malignancies. Although not statistically significant, a slight predominance of solid tumors over hematologic malignancies was observed. This may be partly attributed by the current trend toward more intensive chemotherapy and improved survival in pediatric patients with solid tumors [15]. With regard to primary therapy, all patients enrolled in this study received combinations of alkylating agents and topoisomerase II inhibitors, making it difficult to implicate a single causative agent.
The median latency period until the development of t-MN was 2.4 years, relatively shorter than that observed in previous studies of adult t-MN patients. In a series of 306 patients with t-MNs reported by Smith et al. [3], the median latency was 5.2 years. Three-fourths of our patients were initially diagnosed with overt leukemia without a preceding period of myelodysplasia, and the most frequently observed karyotype was an 11q23 abnormality. Of note, these are all typical findings of t-MNs following treatment with topoisomerase II inhibitors. Our findings suggest that topoisomerase II inhibitors may have a greater impact on the development of t-MNs than alkylating agents in young survivors. Kayser et al. previously showed that a younger age at the time of the diagnosis of the primary disease, as well as the administration of intercalating agents and topoisomerase II inhibitors, were associated with a shorter latency period between the diagnosis of the primary malignancy and the occurrence of t-AML [7]. Our results are also in line with data from a study by Barnard et al. [16], which reported a median survival of 37 months. They also noted that most children with t-MDS/AML had a similar clinical history of exposure to epipodophyllotoxins.
Compared to primary myeloid neoplasms, therapeutic approaches for patients with t-MNs are challenging in most cases. A number of factors explain the dismal prognosis of t-MNs: a high frequency of unfavorable cytogenetic aberrations, persistence of the primary disease, poor hematopoietic reserves, organ dysfunction, and colonization with antibiotic- resistant bacteria and fungi because of a chronic immunosuppressive state. These findings make patients more vulnerable to the acute toxicities of additional myelosuppressive chemotherapy [41718]. The treatment most likely to cure t-MNs is allogeneic HSCT [81920]. Identification of pre-transplantation risk factors is crucial for establishing a therapeutic plan for patients with a t-MN. Although there have been no published pediatric data regarding these risk factors, clues can be drawn from previous adult studies. According to a large adult series published in 2009, age >35 years, poor-risk cytogenetics, inadequate disease control at the time of transplantation, and less well-matched donors were all associated with a poor outcome [20].
This study demonstrated that a significant proportion of children and adolescents with a t-MN could experience a long-term survival if they are treated with a curative intent. Allogeneic HSCT appears to play a key role for attaining a cure in these young patients, even those who fail to achieve CR or lack a matched-related donor. Although patients whose disease was in remission showed a higher survival rate after transplantation, half of those who had persistent disease were salvaged by transplantation.
The major limitations of our study include small patient numbers, data from only a single center, and some missing data for several patients. Together, these limitations precluded an in-depth analysis and made it difficult to draw statistically significant conclusions. Despite the small size of the cohort, our results showed an improved survival outcome compared to previous studies involving children with t-MNs. In a series of 21 children with t-MDS/AML who underwent allogeneic transplantation at St. Jude Children's Research Hospital, Hale et al. [8] reported a 3-year disease- free survival of 19%. Four patients survived, while the remaining patients died of regimen-related toxicity (N=7) or relapse (N=10). In 2006, this report was further extended by Woodard et al. [9], to include a total of 38 patients who underwent allogeneic transplantation. Both the 3-year overall and EFS were 15%, and the 3-year non-relapse mortality rate was 60%. Another more recent report from the MD Anderson Cancer Center described 22 patients with t-MDS/AML among 2,589 children treated for cancer. The 2-year survival rates were 20%, 40%, and 25%, in patients receiving allogeneic HSCT without induction therapy, patients transplanted in remission after AML-type induction therapy, and patients receiving transplantation as salvage therapy, respectively [10].
Several explanations may account for our better survival outcomes compared to previous studies. The studies cited above dealt with relatively old data, the most recent one having been published in 2009 [10]. Recent advances in transplantation techniques and supportive care may have contributed to the improved outcomes observed in this study. Another possible explanation is that most patients in this study had received induction therapy before transplantation. This may have contributed to our improved outcome, by reducing tumor burden in advance. Considering that 4 of the 5 patients who were in CR at the time of transplantation survived, our strategy for reducing tumor burden before transplantation seems effective. A hasty, pessimistic judgment regarding the outcome of t-MNs in pediatric patients should thus be avoided. We strongly suggest that these young patients be considered for allogeneic HSCT with a curative intent.
In conclusion, a significant proportion of children and adolescents with t-MNs can experience a long-term survival, and allogeneic HSCT appears to play a key role for attaining a cure in these young patients. Allogeneic HSCT should be considered for most children and adolescents with t-MNs, as young patients generally exhibit better tolerance to chemotherapy and have fewer co-morbidities compared to adults [21]. Further investigations should be directed toward developing more effective and safer transplantation protocols for the treatment of patients with t-MN.

 XML Download
XML Download