

 PDF
PDF ePub
ePub Citation
Citation Print
Print


INTRODUCTION
Hematopoietic stem and progenitor cells (HSPC) stand at the top of the hematopoietic hierarchy and by a well-balanced interplay between self-renewal and differentiation, they ensure the lifelong supply of mature blood cells. Physiologically, these HSPC reside within the so-called bone marrow (BM) microenvironment which harbors distinct stem cell “niches”. Together with cell-intrinsic mechanisms, these spatial and functional units of the BM microenvironment are essentially involved in the regulation of HPSC behavior. During the last decades, mesenchymal stem or stromal cells (MSC) have been identified as one of the main cellular components of the BM microenvironment holding an indispensable role for normal hematopoiesis [12]. Given the major advances in our understanding of the physiological role of the BM microenvironment and in particular mesenchymal stem and progenitor cell (MSPC), there is also growing interest regarding the contribution of the BM microenvironment to the pathogenesis of myeloid stem cell disorders such as acute myeloid leukemia (AML) and myelodysplastic syndromes (MDS). This review highlights recent work investigating the role of the BM microenvironment in AML and MDS in mice and humans and summarizes the current knowledge with a special focus on MPSC. Furthermore, potential models integrating the BM microenvironment into the pathophysiology of these myeloid disorders and how this crosstalk between clonal hematopoietic cells and altered stem cell niches may be therapeutically targeted in the future will be discussed.
THE HEALTHY BONE MARROW MICROENVIRONMENT
The concept that HPSC require specialized anatomical and regulatory spaces within the BM to maintain their functionality was first postulated by Schofield in 1978 [3]. Following this initial description, tremendous advances in imaging techniques and selective genetic manipulation of specific cell population have facilitated detailed insights into the anatomy and physiology of the BM microenvironment, particularly in mice. Furthermore, these experimental proceedings enabled the identification of key components of the BM cavity and their anatomical and functional interactions.
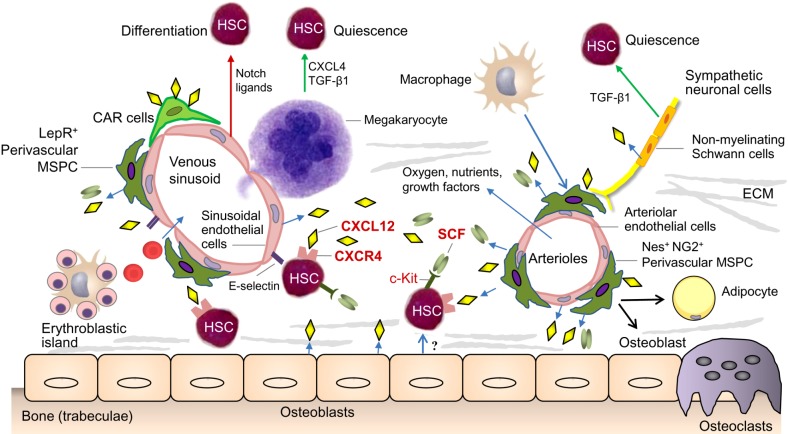
On the one hand, the BM microenvironment consists of non-cellular components like extracellular matrix (ECM) proteins including collagen, fibronectin, laminin and hyaluronic acid. On the other hand, it is mainly composed of cellular elements, which can be categorized into cells exhibiting a hematopoietic origin and non-hematopoietic cells. While the hematopoietic cells are mainly represented by macrophages, megakaryocytes, neutrophils, osteoclasts and regulatory T cells, the non-hematopoietic cells consist of MSPC, osteoprogenitors, osteocytes, endothelial cells, adipocytes, sympathetic nerve fibers as well as non-myelinating Schwann cells (Fig. 1). These niche cells bi-directionally communicate with HSPC via secreted factors including stem cell factor (SCF), transforming growth factor beta-1 (TGF-β1), platelet factor 4 (PF4 also called CXCL4), angiopoietin 1 (ANGPT1) and thrombopoietin (TPO). Sympathetic nerve fibers modulate the circadian egress of HSPC from the BM via the secretion of noradrenaline. Furthermore, chemokines such as stromal cell-derived factor 1 (SDF1 also called CXCL12) and cytokines are also involved. Beside these soluble factors, this crosstalk between surrounding cells and HSPC is also mediated by cell-bound molecules such as adhesion molecules and Notch ligands. Finally, also the oxygen tension within the niches seems to regulate the behavior of HSPC (456).
THE ROLE OF MSPCS IN HEALTHY HEMATOPOIESIS
While there has been a long history of research and debate about the contribution of bone cells to healthy hematopoiesis, the indispensable role of MSPC for the regulation of normal HSPC has primarily been unraveled during the last years. The first description of such mesenchymal precursor cells in the BM goes back to studies of Friedenstein et al. [7], who identified colony-forming units of fibroblasts (CFU-F) obtained from BM suspensions. Besides the stem cell-defining property of self-renewal, these mesenchymal precursor cells exhibit the potential to differentiate into different mesodermal lineages such as bone, cartilage and fat cells. As a consequence and translated from their assumed embryonic counterparts, these cells have been termed ‘mesenchymal stem cells’ during the past. However, also the term ‘mesenchymal stromal cells’ is commonly used to describe these cells. This terminological inaccuracy reveals our incomplete understanding of this cell population and points to its heterogeneity. This cell bulk probably consists of ‘true stem cells’on the one hand, but on the other hand also includes mesodermal cells without self-renewal potential and limited differentiation capacities. In mice, several MSC subsets have been identified based on the expression of specific markers, including Nestin (Nes), neuron/glial antigen 2 (NG2), platelet-derived growth factor receptor alpha (PDGFRA or CD140), CD51, Sca-1 and/or leptin receptor (Lep-R). Furthermore, another MSPC population, the so-called CXCL12-abundant reticular (CAR) cells, is defined by high expression of CXCL12 (Fig. 1). Besides their marker profiles, these subsets differ also in parts with regard to their location within the BM cavity and their functional properties [1246]. In contrast to this advanced understanding of MSC in mice, the knowledge about human MSC is more limited due to technical limitations in tracking and visualizing these cells in vivo and in vitro. To cope with these restrictions and to facilitate as much as possible the comparability of results retrieved from differentially generated cells, the International Society for Cellular Therapy has proposed minimal definition criteria for MSC which are based on surface marker expression, plastic adherence and in-vitro differentiation potential [8]. Although this definition represented a step forward for the standardized research in this field, it has to be acknowledged when interpreting results from different researchers that culture-expanded human MSC are heterogeneous and contain different compositions of true stem cells and more differentiated stromal cells.
PATHOGENESIS OF MDS AND AML: THE ROLE OF MSC
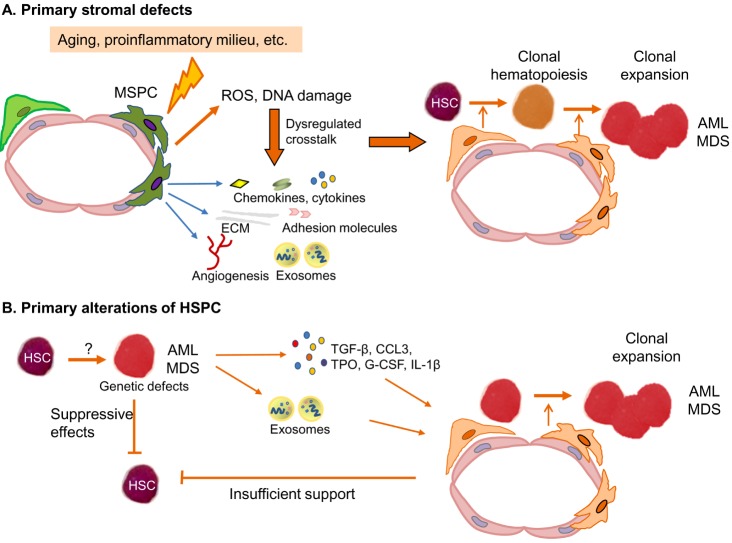
Based on clonal tracking experiments and murine transplantation models, it is a well-accepted theory that MDS and AML originate from HSPC (Fig. 2). Induced by an unknown trigger or potentially as recently shown as a consequence of aging, HSPCs acquire molecular aberrations which confer a survival advantage to them over normal HPSC in the BM. Subsequently, there is a clonal expansion of HSPC which by the acquisition of additional genetic and molecular alterations finally transform into malignant MDS or AML hematopoietic cells [910]. On the one hand, the underlying molecular aberrations of MDS- and AML-derived HPSC relate to genetic lesions such as translocations, inversions, and loss of chromosomal material. On the other hand, DNA mutations of several biological pathways such as DNA methylation machinery, chromatin modification, splicosome, transcription factors and signaling pathways have been unraveled during the last years [111213]. It seems that some mutations such as DNMT3A and TET2 mutations might develop at an early stage of MDS and AML pathogenesis, and other mutations might come up during the course of disease contributing to disease progression [1415]. Furthermore, MDS patients exhibited a median of 3 mutations (range, 0–12) and AML patients a median of 5 mutations, and one specific single mutation alone can induce some MDS-specific or AML features in mice [1112]. This suggests that mainly the interaction between several mutations can probably cause the MDS or AML phenotype. Nevertheless, despite a better understanding of the molecular landscape, the cellular mechanisms by which MDS- and AML-derived HSPC replace normal HSPC in the BM and thereby cause hematopoietic insufficiency, have just been started to get unraveled. These might be mediated by direct interactions between MDS- and AML-derived progenitor cells and healthy HPSC, but also indirectly by interactions with the BM microenvironment, in particular MSPC.
NICHE-INDUCED MYELOID MALIGNANCIES
Taking into account the physiological relevance of the BM microenvironment, several investigators have tested the hypothesis if genetic alterations of niche cells are capable of inducing myeloid malignancies. This idea challenges the well-accepted concept that MDS and AML originate exclusively from HSPC-intrinsic genetic defects.
First evidence for niche-driven myeloid disorders came from a series of murine experiments with deletions of the Retinoblastoma gene, the retinoic acid receptor gamma (RAR-γ), IκBα or the Notch ligand endocytosis regulator Mib1 [16171819]. In all experimental systems, mice developed a myeloproliferative disease phenotype characterized by leukocytosis, extramedullary hematopoiesis and splenomegaly. These studies highlighted that genetic changes of non-hematopoietic elements of the BM microenvironment can directly contribute to disease initiation. Still, in some of these experiments, simultaneous deletion of the candidate gene in hematopoietic and surrounding cells was required thus preventing to dissect the cell subset responsible for the myeloproliferative phenotype.
Two subsequent studies refined this concept and by specific genetic manipulation of specific stromal cells subsets, they pointed towards a pivotal role of MSC and osteoblasts for the pathogenesis of MDS and AML. In a first sophisticated model reported by Raaijmakers et al. [20], genetic ablation of the RNAse III endonuclease Dicer1 in mesenchymal/osteoprogenitor cells resulted in a MDS phenotype associated with acquisition of genetic alterations and a propensity to develop AML. In this model, loss of Dicer1 resulted in a lower expression of the SBDS gene, which is involved in ribosomal maturation and mutation in human Shwachman-Bodian-Diamond syndrome (SBDS), a congenital BM failure disorder associated with leukemic predisposition. Of particular interest, reduced expression of Dicer1 and the SBDS gene has also been described in MSC from patients with MDS suggesting that some pathophysiological aspects from murine experiments maybe also apply to human MDS [212223]. In a recent study by Zambetti et al. [24], there was a further support of the theory that the stromal compartment is actively involved in the initiation and propagation of myeloid diseases. Here, deletion of the SBDS gene again in mesenchymal progenitor cells induced mitochondrial dysfunction, oxidative and genotoxic stress in HSPC. This direct link between a stromal defect and subsequent disturbances of HSPC functionality was mechanistically mediated by damage-associated molecular pattern (DAMP) molecules S100A8 and S100A9, which were secreted from mesenchymal niche cells. Potentially relating this finding also to human MDS pathogenesis, the authors detected a significant association with between the expression of S100A8 and S100A9 and the likelihood of AML transformation in 45 patients with low-risk MDS. Furthermore, the role of the innate immune toll-like receptor (TLR) signaling in the pathogenesis of human MDS has just recently been described by two other groups [252627].
In a second model, constitutively active β-catenin and subsequent Wnt signaling in osteoblasts caused a transplantable AML phenotype [28]. These changes were induced by the expression of Jagged-1 in niche cells and consequently activation of Notch signaling in hematopoietic cells. Again, translating this into humans, these authors found increased nuclear β-catenin in osteoblastic cells and increased Notch activity in HSPC of 38% of patients with AML or MDS. Consistent with this, we also previously reported on the increased expression of Jagged-1 on MSC derived from patients with MDS and AML [2129].
MALIGNANT MYELOID PROGENITOR CELLS MODULATE THE BM MICROENVIRONMENT
There are also several examples illustrating the second major concept that malignant HSPC remodel the bone marrow microenvironment. In a murine model of blast-crisis, CML osteoblasts were significantly reduced and functionally inhibited by leukemic cells. This suppression of bone formation was amongst others measurable by decreased levels of osteocalcin, a finding that we also found in humans with newly-diagnosed AML. This functional inhibition of osteoblasts in this model was mainly mediated by CCL3 (also known as macrophage inflammatory protein 1α, MIP-1α) [30].
Furthermore, using a mouse model of chronic phase CML, two studies also demonstrated how leukemic cells directly alter surrounding niche cells [3132]. In the first model, secretion of G-CSF by leukemic cells resulted in decreased expression of CXCL12 by BM stromal cells and hereby impaired HSPC maintenance. In the second model, CML cells stimulated the expansion of osteoblasts by the secretion of thrombopoietin (TPO) and CCL3, and direct cell-cell contact between leukemic cells and MSC. These osteoblasts exhibited reduced expression of HSCP retention factors and impaired capacity to support normal hematopoiesis. In contrast, these niche alterations had no major impact on the behavior of the leukemic cells, probably to their altered susceptibility to signals from the BM microenvironment. Furthermore, TGF-β, Notch and inflammatory signaling were responsible for the development of fibrosis in this model.
Besides this direct modulation of mesenchymal cells through leukemic signals, two recent studies pointed at nerve cells as potential target for leukemia-associated remodeling of the BM microenvironment.
In a JAK2V617F MPN mouse model and in a MLL-AF9 AML transplant model, invasion of leukemic cells caused direct damage of nerve cells subsequently affecting the functionality of MSC [3334]. In the former model, this neuropathic effect on sympathetic fibers and ensheating Schwann cells was mediated via the secretion of IL-1β from malignant cells. This effect favored an acceleration of a myeloproliferation counterbalanced by an inhibition of normal hematopoiesis and could be reversed by treatment with β3 adrenergic agonists.
Taken together, all of these data suggest that the leukemia-induced manipulation of niche elements probably serves as a self-reinforcing mechanism to support leukemic cells at the expense of normal hematopoiesis (Fig. 2).
ALTERATIONS OF MSPC IN PATIENTS WITH MDS AND AML
These above mentioned findings were mostly retrieved from animal models and in-vitro systems and therefore raise the question about the contribution of MSC to the pathogenesis of human AML and MDS. Unfortunately, in contrast to HSPC, MSC had been studied in considerably less detail for a long time, which initially created controversies regarding their contribution to MDS and AML pathogenesis.
In addition to the changes of MSC already mentioned in the chapters above, genetic aberrations, different from those in hematopoietic cells, can be reproducibly found in approximately 25% of MDS- and AML-derived MSC [35]. Furthermore, several cytokines, adhesion molecules and transcription factors have been described to be altered in MDS- and AML-derived MSC, but it is unclear whether and how these abnormalities influence the pathogenesis of MDS and AML. Conflicting results have been also reported with regard to the biological behavior of MDS- and AML-derived MSC. Although their clonogenic potential and hematopoietic support capacities were shown to be reduced in some MDS and AML patients, other data suggested a proliferative advantage and normal hematopoietic support by MDS- and AML-derived MSC. These controversies were probably related to a limited number of samples as well as different experimental settings [363738394041424344454647484950515253].
By investigating the largest groups of patient-derived MSC samples so far we have shown, that MSC across all MDS subtypes as well as from de novo AML were structurally, epigenetically and functionally altered, thus causing impaired stromal support of HSPC [2129]. These findings have been recently confirmed by results from another group [54] and clearly indicate that impaired stromal support substantially contributes to hematopoietic insufficiency in MDS and AML.
Overall, these data demonstrate that the BM microenvironment and in particular MSC are also involved in the pathophysiology of human AML and MDS. Furthermore, it has opened a new direction to investigate the biology of these two myeloid malignancies and may help to unravel new therapeutic approaches.
WHO COMES FIRST IN THE PATHOGENESIS OF AML AND MDS: HSPC OR MSC?
This categorical question whether AML and MDS derive from primary defects of HSPC or whether stromal disturbances stand at the beginning of the pathogenesis cannot be answered. As summarized above, there are evidences from mice and humans that both concepts contribute to the development of AML and MDS as well as the associated hematopoietic insufficiency. This concept of bidirectional interaction between MSPC and HSPC in MDS has been nicely demonstrated by Medyouf et al. [55]. In their xenograft transplant model, HSPC from human low-risk MDS only engrafted when co-transplanted with autologous MSPC indicating their dependency on deregulated signals from MSPC. On the other hand, after exposure to hematopoietic MDS cells, healthy MSPC adopted a phenotype resembling that of primary MDS-derived MSPC. In line with this idea that malignant HSPC actively modulate their niche, we recently showed that supernatants from AML cell lines induced changes in healthy MSPC comparable to those observed in primary AML-derived MSPC [29].
Personally, we assume that primary stromal alterations may preferentially occur in the initiation phase of such diseases (Fig. 2A). Potentially as a consequence of aging or previous therapies, stromal changes may actively modulate HPSC functions via inflammatory signals hereby enabling the acquisition of genetic defects. This may contribute either to the development or the establishment of clonal hematopoiesis. Still, we also believe that a single stromal aberration, as shown for example by deletion of Dicer-1 in mice [20], is probably not sufficient alone to induce the phenotypic heterogeneity of human AML and MDS. However, stromal alterations might play a relevant role for the rare phenomenon of donor-derived leukemia after allogeneic blood stem cell transplantation [56]. This might also be the case in germline mutations associated with inherited myeloid malignancies. Here, also stromal and hematopoietic cells are equally affected by the predisposing genetic defect and may therefore also equally contribute to disease manifestation [57].
At later phases of the disease, when the bone marrow is dominated by malignant HSPC, based on the available literature, these hematopoietic cells actively modify the surrounding microenvironment resulting in disease propagation and suppression of normal hematopoiesis. In this context, it also needs to be noted that besides manipulating niche elements, leukemic and MDS cells also directly suppress healthy hematopoietic cells, for example by exosome transfer.
Independent from this question regarding the cellular origin, the next step to move this idea forward will be to elucidate the communication pathways between stromal and hematopoietic cells. This may enable to develop stromal-targeted therapies aiming to eradicate malignant hematopoietic cells and to restore normal hematopoiesis.
TARGETING THE BM MICROENVIRONMENT IN AML AND MDS
The growing knowledge about the niche-mediated effects in the pathogenesis of myeloid disorders begs the question whether these interactions can be therapeutically targeted. In contrast to currently available treatment options like classical chemotherapy or targeted therapies (e.g. tyrosine kinase inhibitors), such a stromal-directed therapy would represent a novel non-cell-autonomous approach. Hereby, it could synergistically act with conventional therapies, and might overcome resistance mechanisms and would not be restricted to patients with the specific molecular aberration (e.g. FLT3-ITD).
In a first attempt, several investigations, that are mainly preclinical but also already some early clinical, aim to reverse the resistance to chemotherapy and tyrosine kinase inhibitors by interfering the adhesion of leukemic cells to stromal cells. In this context, the CXCR4-CXCL12 axis, VLA-4, E-selectin and CD44 have been identified as interesting candidates for such an approach [585960616263]. Although combining this with conventional therapies has already demonstrated synergistic effect in the preclinical setting, limited results from studies in humans are available yet [64]. These do not allow a definitive estimation of the efficacy of this strategy so far.
Furthermore, there is a hint that the immunomodulatory compound Lenalidomide, which is approved for the treatment of MDS patients with isolated deletion 5q (del5q), may mediate in parts its effect through modulation of the BM microenvironment [65]. This mechanism may also be conceivable for the second treatment approved for the treatment of MDS in Europe, namely the hypomethylating agent Azacitidine. Indeed, we recently described that MDS- and AML-derived MSC exhibit a specific epigenetic signature, suggesting that its reversal may contribute to the therapeutic effects of Azacitidine in patients with MDS [2129]. Finally, blocking of ligands of the TGF-β superfamily such as GDF-11 and others by ligand-traps such as Luspatercept and Sotacerpt is a new concept to treat anemia in low-risk MDS patients and is currently under investigation. Again, it might be the case that some proportion of the efficacy of these drugs is related to the modulation of the BM microenvironment [6667].
CONCLUSIONS
A better understanding of the physiological role and composition of the BM microenvironment has stimulated the interest of the scientific community to investigate its contribution to the pathogenesis of myeloid disorders. Results in particular from preclinical mouse models and to a certain extent from human samples support this idea. Current research initiatives aim to decipher the bi-directional crosstalk between malignant hematopoietic cells, niche cells and normal HSPC. Optimally, this will help to identify new stromal-directed strategies for the treatment of AML and MDS.
 XML Download
XML Download