

 PDF
PDF ePub
ePub Citation
Citation Print
Print


TO THE EDITOR: Delta beta (δβ)-thalassemia results from a deletion in both the delta and beta genes on chromosome 11. The gamma genes on the affected chromosome increase their production of gamma globin, thereby increasing the amount of hemoglobin F (HbF). δβ-Thalassemia heterozygotes clinically show characteristics of thalassemia minor. However, homozygous δβ-thalassemia may give a clinical picture of thalassemia intermedia with a mild anemia.
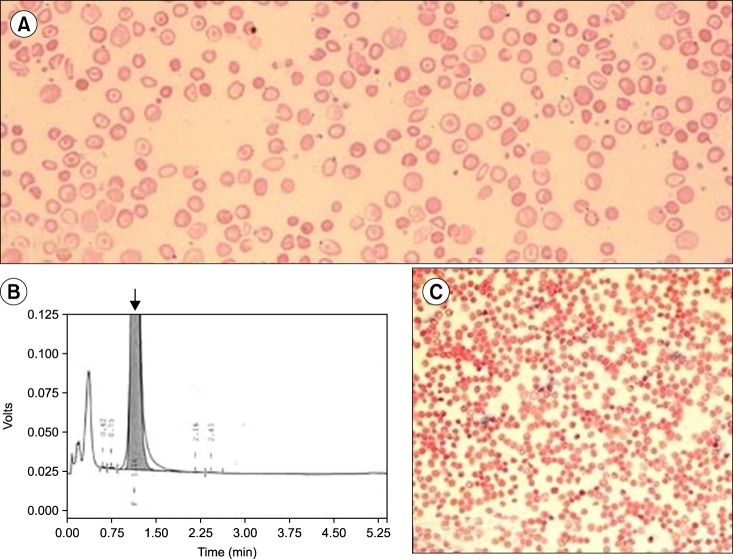
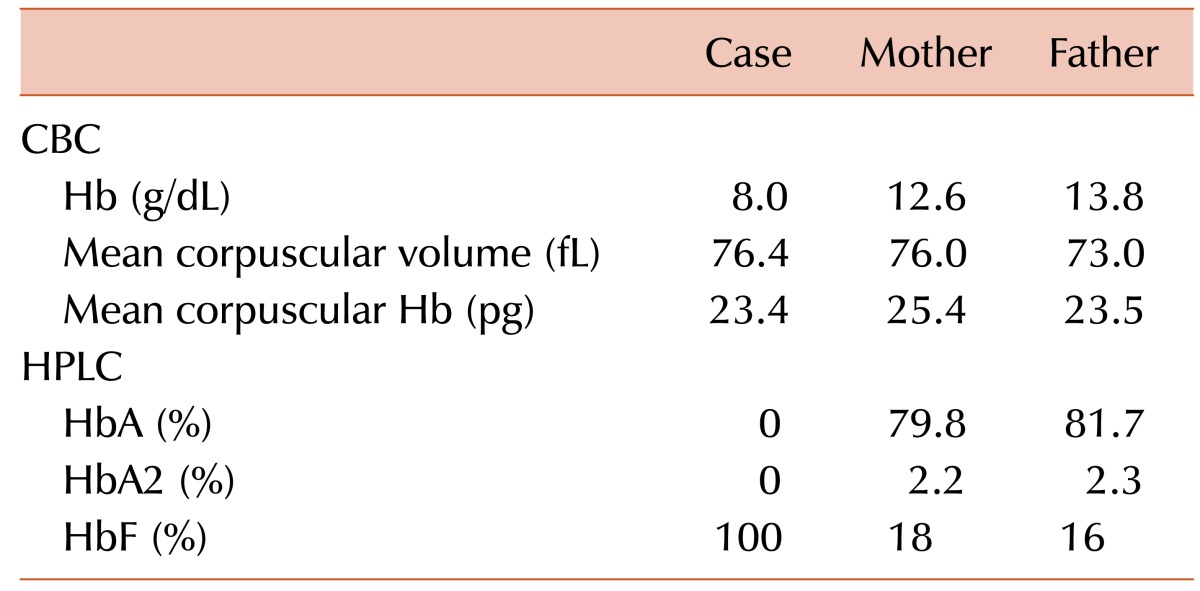
A 12-month-old boy presented to the hematology outpatient department for evaluation of pallor and jaundice that had been for the past 2 months. He had no history of a blood transfusion. His family history was insignificant for congenital anemia. His parents had a consanguineous marriage. Physical examination revealed pallor and palpable spleen 2 cm below the left costal margin. Other examination findings were unremarkable. A complete blood count (CBC) revealed an Hb level of 8.0 g/dL, WBC count of 8.9×109/L, and platelet count of 341×109/L (Table 1). A peripheral blood smear revealed anisopoikilocytosis with hypochromic microcytic red cells, target cells, and basophilic stippling (Fig. 1A). The corrected reticulocyte count was 1.6%. Liver function tests showed raised levels of serum total bilirubin (3.5 mg/dL) and indirect bilirubin (3.0 mg/dL). High-performance liquid chromatography (HPLC) showed 100% HbF and an absence of HbA and HbA2 (Fig. 1B). The Kleihauer-Bekte test revealed a pancellular pattern (Fig. 1C). Consequently, a CBC followed by HPLC was performed for both parents who were apparently healthy and had no history of blood transfusions (Table 1). Kleihauer-Betke tests of both parents showed a heterocellular distribution of HbF.
Hence, the patient was diagnosed with homozygous δβ-thalassemia, whereas the parents with heterozygous δβ-thalassemia. Unfortunately, mutational analysis could not be performed because the patient was lost to follow-up.
δβ-Thalassemia results from the deletion of both δ and βgenes. Homozygotes for δβ-thalassemia have 100% HbF and, because of the increased synthesis of HbF, may have thalassemia intermedia rather than thalassemia major [1].
However, the phenotype of heterozygotes resembles that of the β-thalassemia trait, but the HbA2 percentage is not increased and is often normal. HbF in such individuals is consistently elevated, varying from 5% to 20%. Peripheral blood film findings are similar to those for the β-thalassemia trait, and the distribution of HbF is heterocellular, which is best observed via flow cytometry. It is necessary to distinguish it from hereditary persistence of fetal hemoglobin (HPFH). The two groups of disorders are distinguished by the phenotype of heterozygous individuals. Heterozygotes of δβ-thalassemia mutations have 5% to 20% HbF, which is heterocellularly distributed in red cells, whereas heterozygotes of HPFH mutations have 17% to 30% HbF, with a pancellular distribution. In addition, homozygotes of HPFH are asymptomatic, whereas δβ-thalassemic homozygotes have thalassemia intermedia-like features [2].
At least nine mutations can result in δβ-thalassemia. This type of thalassemia is observed in many ethnic groups, including some Mediterranean populations (Italians, Greeks, and Turks). Although the exact diagnosis of δβ-thalassemia requires genetic analysis for mutations, Hb electrophoresis or HPLC findings of markedly elevated HbF may be suggestive. An extensive PubMed search was done to determine the incidence of δβ-thalassemia in different parts of the world, but owing to the rarity of this Hb variant, only a handful of case reports were identified from across the world [3456].
 XML Download
XML Download