

 PDF
PDF ePub
ePub Citation
Citation Print
Print


INTRODUCTION
Mast cell leukemia (MCL) is the most aggressive form of systemic mastocytosis (SM). Common symptoms of MCL include flushes, fever, malaise, diarrhea, and tachycardia. Diagnosis of MCL requires that (i) SM criteria are fulfilled, and (ii) bone marrow (BM) atypical mast cells (MCs) comprise ≥20% of the total white blood cells (WBCs). MCL is composed of a leukemic variant (>10% in the peripheral blood) and an aleukemic variant (<10% in the peripheral blood). This orphan disease accounts for less than 1% of all SMs [1].
Owing to its rarity, the pathogenesis and standard treatment for MCL are not well established. According to Georgin-Lavialle et al., most patients with MCL (83%) show a normal karyotype in conventional cytogenetic exams. At the molecular level, mutations in the KIT gene have been well investigated in MCL patients, even before the next-generation sequencing (NGS) era. A few therapeutic options for MCL are available, but no efficient treatment has been reproducibly validated. Thus, well-organized clinical trials should be primarily considered [1]. Without proper clinical trials, patients with mutations in KIT other than D816V or those with wild type KIT could be treated with an ABL kinase inhibitor, such as imatinib. Unfortunately, ABL kinase inhibitors are not effective in patients with the KIT D816V mutation. Midostaurin, a multi-target protein kinase inhibitor, can be administered to patients with MCL regardless of mutations in KIT. Allogeneic stem cell transplantation may be a potential curative option for MCL, but a retrospective study found that the response rate was low (three-year survival rate=17% [2 of 12]) [2]. Traditionally, polychemotherapy, such as an AML-type induction regimen, has been used for cytoreductive therapy. Alternatively, steroids and and interferon-α be considered as treatment options.
With the exception of the KIT gene, molecular study and targeted therapy have not been thoroughly evaluated. Therefore, we attempted to treat a refractory MCL patient based on whole exome sequencing (WES) and whole transcriptome sequencing (WTS) of the patient's own DNA and RNA.
MATERIALS AND METHODS
An 18-year-old Korean female visited the Seoul National University Hospital with recurrent pain in the abdomen and both legs that lasted for 1 month. X-rays of the legs and an abdominal computed tomography (CT) scan were performed to determine the cause of the pain and revealed hepatomegaly with ascites and left inguinal lymphadenopathy (largest diameter: 2.4 cm). Excisional inguinal node biopsy revealed dense infiltrates of atypical MCs with strong C-KIT expression; however, no MCs were detected in the peripheral blood (white blood cell count: 7,570/L; 74.9% neutrophils, 21.0% lymphocytes, 3.6% monocytes, 0.4% eosinophils, and 0.1% basophils). We found an increase in immature MCs (24.1%) with bi-lobed nuclei in a BM smear. Most of the MCs showed atypical morphology. No morphological evidence of an associated hematopoietic non-mast cell lineage disease was found. The serum tryptase level was 425.0 µg/L. Chromosomal analysis showed a normal karyotype (46, XX [20]). In addition, we did not observe the KIT D816V mutation. One major and two minor criteria of SM established by the WHO were fulfilled. With the presence of 20% MCs on the BM smear, the patient was diagnosed with the aleukemic variant of acute MCL [3]. Liver dysfuncion was identified as a C finding of the disorder.
We initiated treatment with cytarabine (100 mg/m2) for 7 days and idarubicin (12 mg/m2) for 3 days, but the follow-up BM smear revealed persistence of MCL (MCs: 5.5% of total nucleated cells). Because the patient was reluctant to undergo allogeneic stem cell transplantation, we performed WES and WTS to find druggable mutations or activated signaling pathways.
Next-generation sequencing
BM blasts were acquired at diagnosis, and epithelial cells from saliva were obtained after induction chemotherapy. We used genomic DNA purification kits (Norgen Biotek Corp, Thorold, ON, Canada) to isolate the DNA. Quality was monitored by the NGS QC Toolkit (National Institute of Plant Genome Research, New Delhi, India). DNA was then fragmented for massively parallel sequencing via the HiSeq 2000 system (Illumina Inc., San Diego, CA, USA) according to the manufacturer's instructions. We used the SureSelect Human All Exon Kit (Agilent Technologies Inc., Santa Clara, CA, USA) for DNA capture. FASTQ files were aligned to the UCSC human reference genome (build hg19) using the Burrows-Wheeler Aligner (bwa-0.7.5a) [4] to generate a sequence alignment/map file. Next, the Genome Analysis Toolkit (GATK; Broad Institute, Cambridge, MA, USA) was used for local alignment [5]. Single nucleotide variants (SNVs) and small insertions and deletions (indels) were identified using GATK. SNVs were also identified using MuTect software (Broad Institute) [6]. CONTRA was used to determine copy number variations [7].
For RNA preparation, total RNA quality was assessed using the NanoDrop1000 spectrometer (Thermo Scientific, Wilmington, DE, USA). We used the TruSeq RNA library preparation kit for RNA isolation (Illumina, San Diego, USA). In brief, messenger RNA (mRNA) was purified using polyA selection, then chemically fragmented and converted into single-stranded cDNA using random hexamer priming. Next, the complimentary strand was generated to create double-stranded cDNA (ds-cDNA) that could be used for TruSeq library construction. The short ds-cDNA fragments were then connected with sequencing adapters, and suitable fragments were separated by agarose gel electrophoresis. Finally, TruSeq RNA libraries were built by PCR amplification, quantified using qPCR according to the qPCR Quantification Protocol Guide, qualified using the Agilent Technologies 2100 Bioanalyzer (Agilent Technologies, Palo Alto CA, USA), and then sequenced using the HiSe 2000 platform (Illumina, San Diego, USA). The sequencing data were aligned to genecode v18 by Tophat v1.0.12 (Tophat, genecode reference). Raw counts of mRNA were quantified using Samtools [8], and corresponding reads per kilobase per million reads (RPKM) were calculated using an in-house R script. We used RPKM value to determine differentially expressed genes. Fusion genes were analyzed by FusionMap [9]. To find upregulated intracellular signaling pathways, we used the Gene Set Enrichment Analysis (http://www.broadinstitute.org/gsea/msigdb/annotate.jsp, Broad Institute, MA).
RESULTS
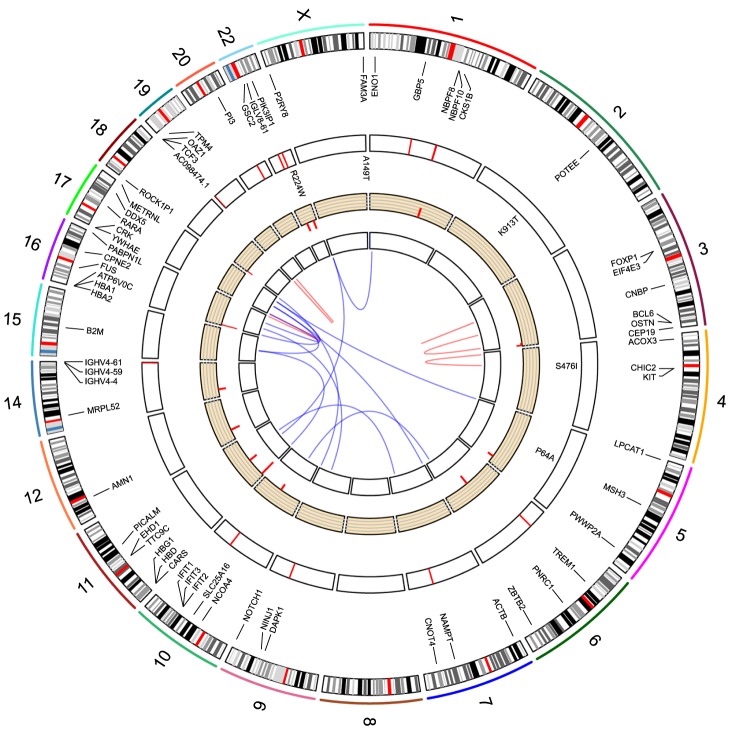
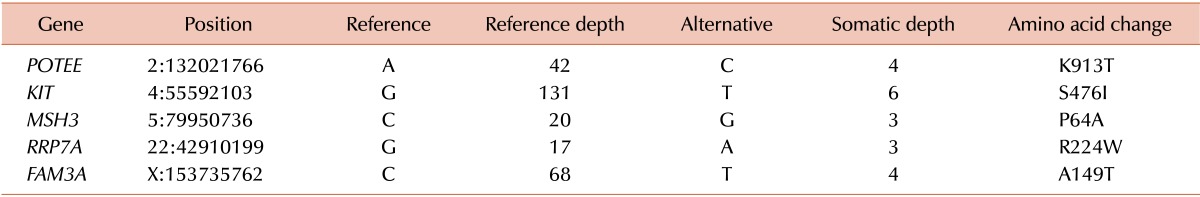
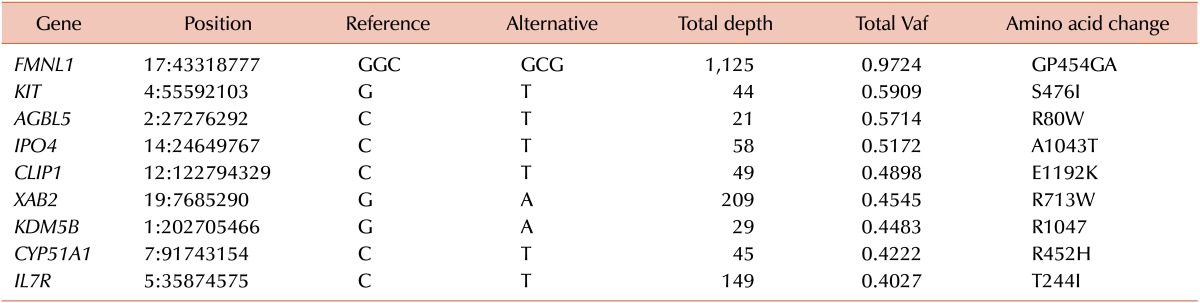
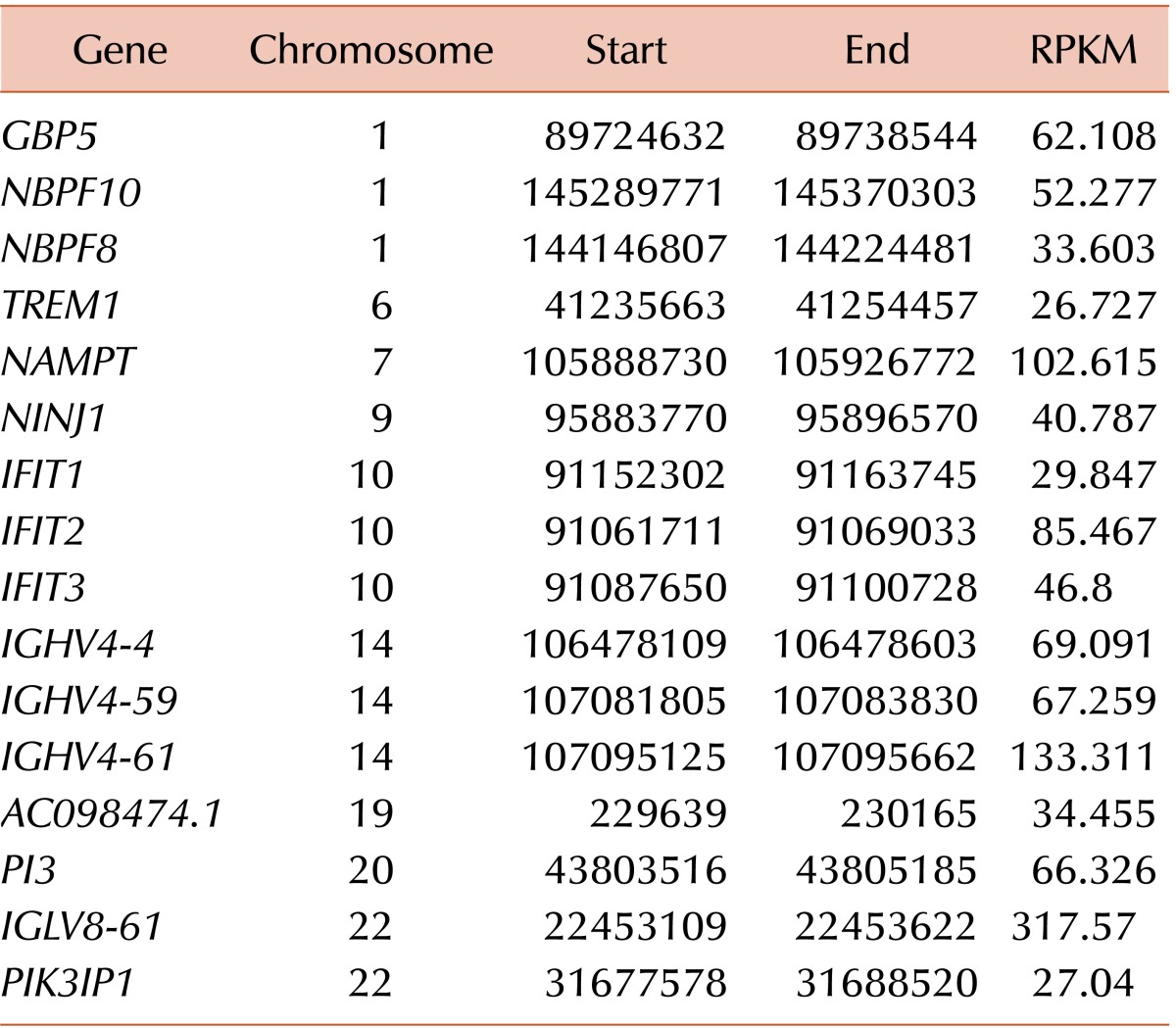
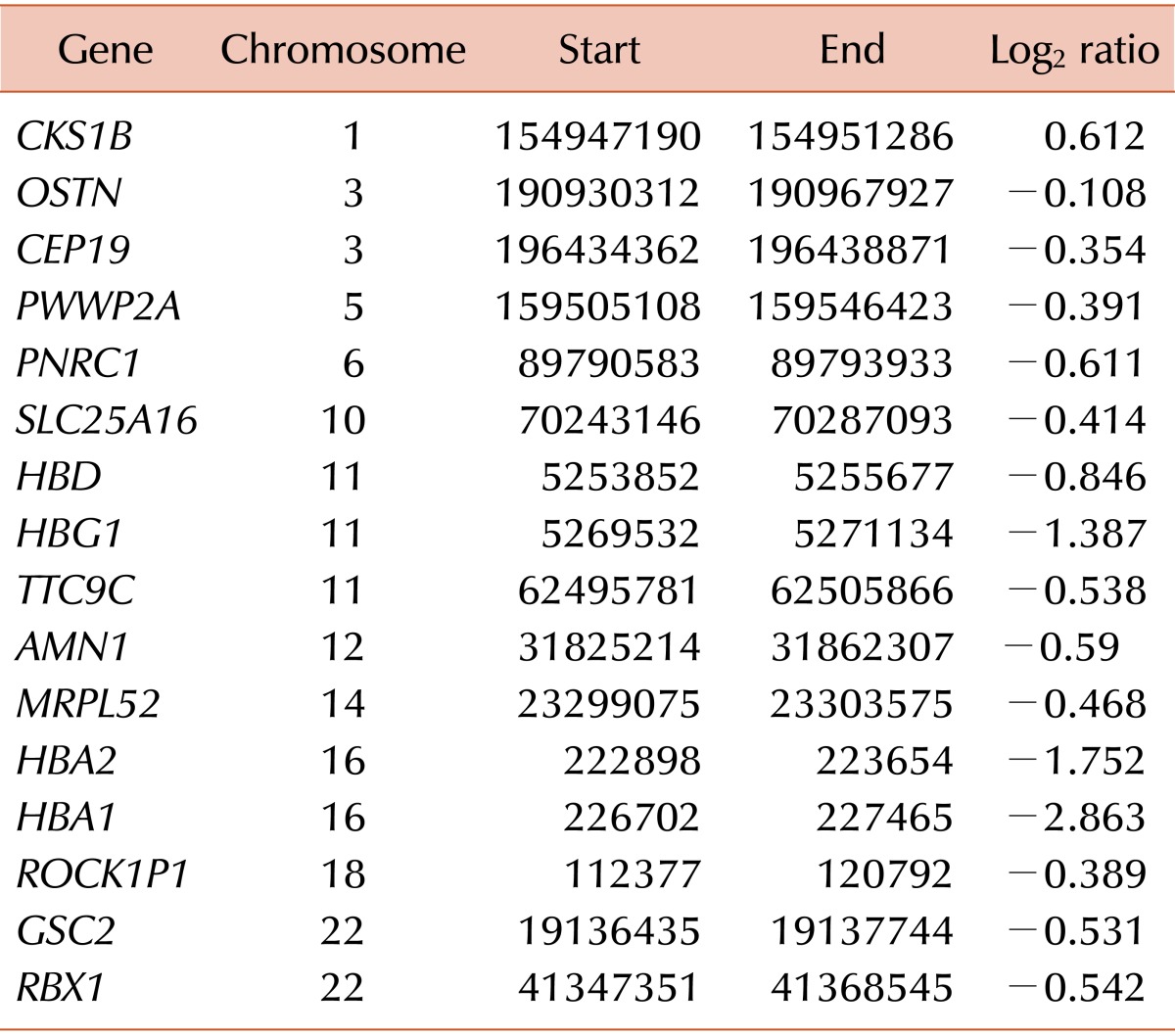
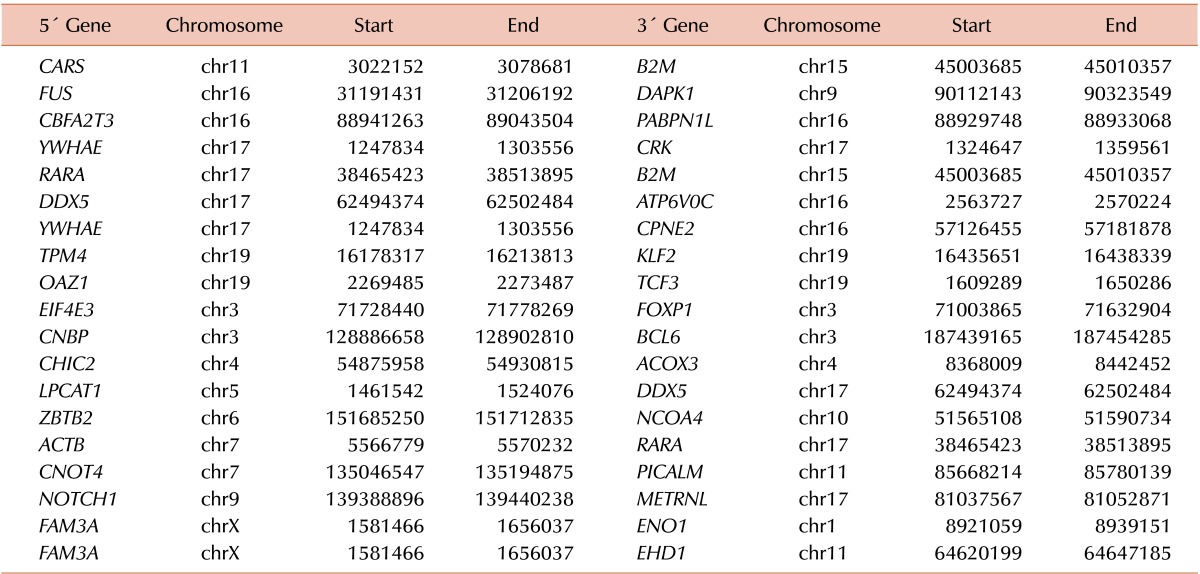
WES analysis yielded a total of 226,007,918 mapped reads (BM DNA: 103,545,760; salivary epithelial DNA: 122,462,158). The mean read depth of the neoplasm was more than 100-fold. WES analysis using GATK failed to demonstrate noticeable nonsynonymous SNVs or small indels, whereas MuTect detected five SNVs, including KIT S476I (Table 1). Druggable genes associated with copy number changes were also not found. WTS analysis also found the KIT S476I mutation (Table 2). The possibility of RARα-B2M and RARα-ACTB fusion have been noted. When performing RNA expression analysis with the WTS data, we observed an upregulation of the PI3K/AKT pathway, which is downstream of KIT and the mammalian target of rapamycin (mTOR). Fig. 1 presents a circus plot of the genetic profile of MCL in this patient. The list of genes in the circus plot can be found in Table 1 and Tables 3, 4, 5.
Based on our WES and WTS results, we first administered treatment with all-trans retinoic acid (ATRA), which targets RARα. After 2 weeks of ATRA, the patient's left eye protruded. An orbital CT scan showed the presence of a retro-orbital mass lesion, probably chloroma. Because ATRA failed to demonstrate efficacy, the patient was treated with dasatinib, which targets KIT. Her leg and abdominal pain improved transiently, but worsened after one month. Suspecting disease progression, we stopped dasatinib treatment and began treatment with everolimus, which targets the mTOR pathway. At this time, we also requested a PI3K inhibitor from a pharmaceutical company. Two weeks after beginning everolimus treatment, neutropenia and thrombocytopenia became prominent, probably related to adverse effects of the drug. Despite the everolimus treatment, the patient's bone pain did not improve. Unfortunately, a sudden cardiac arrest occurred 4 weeks later while waiting for the PI3K inhibitor as an alternative drug. She survived 11 months after the diagnosis of MCL.
DISCUSSION
Before our current study, only one WES study in a patient with MCL patient had been published in 2012 [10]. This study detected a point mutation in IgE mast-cell receptor β chain and KIT, but the treatment approach was not reported. With the exception of that WES study, most other MCL studies have focused on KIT mutations. KIT D816V mutation was detected in more than 90% of patients with an SM. Almost 40% of de novo patients with MCL had the KIT D816V mutation [1]. In addition, S476I, F522C, V654A, V560G, duplication of amino acids 501–502 and 502–503, and deletion of amino acids 501–502 were also reported in a case report [101112131415]. Besides the KIT mutation, a TET2 mutation has been investigated in aggressive SM [16]. Few patients with MCL had a 5q deletion, but all of those patients had secondary MCL that evolved from myelodysplastic syndrome [17].
Notably, the KIT mutation is the most important and prevalent pro-oncogenic mutation in MCL. In this study, we found the presence of a KIT S476I mutation, which has been discovered previously in chronic MCL [15]. With our finding, this point mutation becomes the second most common point mutation in MCL, following KIT D816V, which is located in the tyrosine kinase domain. KIT S476I, on the other hand, is located in the immunoglobulin-like extracellular domain, the function of which is not yet known [18]. A small portion of patients with MCL show improvement upon treatment with KIT inhibitors, such as imatinib or dasatinib, but dasatinib was not effective in our patient. Because a few reports showed promising results of PKC412 (midostaurin) for aggressive SM and MCL, we considered administering PKC412 to the patient [192021]. However, the drug was not available at that time in Korea. Recently, selective inhibitors targeting specific KIT mutations, such as KIT D816V, have been studied [22], and selective drugs that block KIT S476I could be beneficial to a proportion of MCL patients. Because no selective KIT inhibitor for S476I is approved currently, we blocked pathways downstream of KIT. One of the downstream pathways is the PI3K/AKT/mTOR pathway, which was upregulated in this MCL case. However, treatment with an mTOR inhibitor regrettably was not effective in this patient.
Several plausible explanations could account for the treatment failure. One explanation would be targeted therapy alone. In patients with Philadelphia chromosome-positive acute lymphoblastic leukemia (ALL), BCR-ABL tyrosine kinase inhibitor (TKI) was used as an induction treatment combined with cytotoxic multiagent chemotherapy, although BCR-ABL TKI alone was very effective in chronic myeloid leukemia [23]. Like ALL, acute MCL, one form of acute leukemia, might be too aggressive to treat with TKI alone. Another explanation could be that KIT S476I, the PI3K/AKT/mTOR pathway, or RARα fusion protein was not a main driver mutation or pathway in this MCL pathogenesis. Although the above variations were detected in sequencing data, our analysis did not exclude the possibility that they were only passenger mutations. Finally, it is possible that the drug dosage level was insufficient, given that the optimal dosage of TKI agents has not been determined for acute MCL.
In conclusion, we presented a case study of a patient with an orphan disease in which we used a targeted approach to therapy with WES and WTS data from the patient. Although this approach did not successfully cure the disease, she survived 11 months, approximately twice as long as the median survival of patients with acute MCL. The results of our treatments were not ideal, but the utility of this type of approach should be further researched and validated in the future.
 XML Download
XML Download