

 PDF
PDF ePub
ePub Citation
Citation Print
Print


INTRODUCTION
Since its discovery in the late 19th century by Röntgen, ionizing radiation has been utilized as one of the three (surgery, chemotherapy, and radiotherapy) most important treatment modalities for many types of cancers [1]. Ionizing radiation kills cells by inducing DNA damage, particularly DNA double-strand breaks, resulting from ionizations in or very close to the DNA [2]. Radiotherapy delivers radiation dose at a typical daily dose of around 2 Gy per fraction, 5 times a week, and up to three weeks, in a regimen called 'fractionated irradiation' [3]. Why use fractionated radiotherapy? This is because the dose that can be delivered to patients is largely limited by their normal tissue toxicity. With the lack of dose delivery technology capable of limiting normal tissue exposure, normal tissue volumes in the conventional radiotherapy have been typically much larger than the tumor volume itself [3]. Hence, the only option for delivering high tumor dose was to use fractionated irradiation regimen.
Latest technological advancement in radiotherapy including stereotactic radiosurgery (SRS) for the brain and stereotactic ablative radiotherapy (SABR) for the extracranial tissues can now deliver individual ablative high doses of radiation (15–24 Gy) to the tumor volume with a very steep dose gradient using highly conformal techniques [1]. Major advantage this technology has brought in the field of radiation oncology is the superior clinical response while significantly lowering normal tissue toxicity, due to the precise targeting ability sparing large volumes of the normal tissues [1]. However, tumors such as glioblastoma multiforme and lung cancers invariably recur and they often do so within the previously irradiated field [456].
How could tumors recur after such ablative doses of radiotherapy? Cancer cells, of course, may bear mutations in many genes, some of which are involved in intrinsic radiation sensitivity, for example, epidermal growth factor receptor (EGFR) and DNA-dependent protine kinase catalytic subunit (DNA-PKcs) [789]. Some cancers may also harbor mutations in apoptotic genes including Tumor protein 53 (TP53) or BCL2 associated X protein (BAX) which can certainly affect the tumor response to irradiation [71011]. Recently, we and others have reported that there are circulating cells, especially those bone marrow-derived cells such as myeloid cells, which can modulate the tumor response to radiotherapy [12]. In this review, we will discuss the mechanisms by which ionizing radiation induce immune responses. More specifically, anti-tumor immune responses, which should be able to bring superior clinical responses, will be summarized. Then, it will be dicussed how tumor microenvironmental factors interferes this anti-tumor immune responses.
INTERPLAY BETWEEN RADIATION AND IMMUNE RESPONSES
Anti-tumor immune responses
Ionizing radiation kills cancer cells by various mechanisms of cell death, including apoptosis, necrosis, mitotic catastrophe, and immunogenic cell death [131415161718]. It has been demonstrated that both single high dose irradiation and fractionated low dose irradiation to tumors lead to the induction of damage-associated molecular pattern (DAMP) molecules [19], including high mobility group protein box 1 (HMGB1), adenosine triphosphate (ATP), heat-shock proteins (HSPs), uric acid, and interleukin-1α (IL-1α) [20] (Fig. 1). In theory, dendritic cells (DCs) should be able to take up these antigens for priming naïve T cells [21]. Furthermore, some irradiated tumors such as breast cancers have shown to release increased levels of granulocyte-macrophage colony-stimulating factor (GM-CSF) [22], the cytokine that induces DC differentiation from monocytes and hematopoietic progenitor cells (HPCs) [23]. In fact, some studies have shown that radiation facilitates DC maturation and migration [24] and an increase in tumor-reactive T cells [25] at ablative irradiation doses of 15–20 Gy. These results do suggest that ionizing radiation should bring a potent anti-tumor response. But in reality how could these anti-tumor immunity fail to trigger a potent anti-tumor response in some patients?
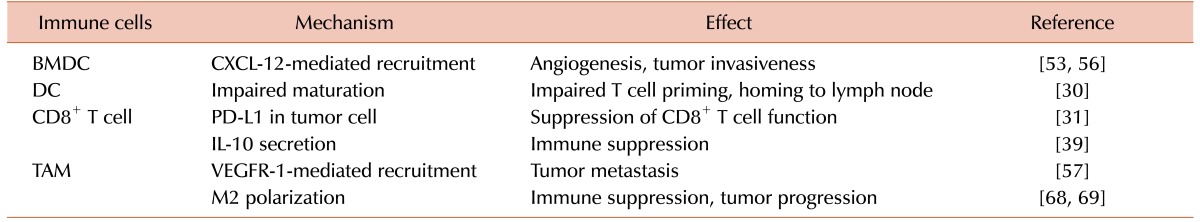
Generation of tumor-specific CD8+ T cells generally requires maturation of DCs capable of antigen uptake and presentation [132627]. This indicates that DC maturation may be severely impaired in cancer patients. It has been extensively demonstrated that tumor hypoxia is a common feature existing in many, if not all, of human and murine solid tumors [28]. Thomlinson and Gray first proposed some 50 years ago a stream of necrotic cancer cells away from the functional blood vessels in histology sections [2]. Later, Brown proposed that fluctuation in the tumor blood flow may cause temporary conditions of low oxygen tensions, leading to acute hypoxic conditions [29]. Tumor hypoxia is well known to be a major hurdle for most anti-cancer drugs because of several reasons: hypoxic cancer cells are far away from blood vessels lowering the anti-cancer drug concentrations to be delivered to those hypoxic tumor cells; hypoxic cells proliferate much slower than well-oxygenated cells escaping the cytotoxic action of many conventional anti-cancer drugs that target rapidly proliferating cells; and hypoxia acts as a selective pressure for more mutations, for example selecting cells that have lost p53-mediated apoptosis [2]. In the perspective of the effect of tumor hypoxia on the anti-tumor immunity, hypoxia is known to inhibit expression of many differentiation and maturation markers including CD1α, CD40, CD80, CD83, CD86, and major histocompatibility complex (MHC) class II molecules in response to lipopolysaccharide (LPS), the stimulatory capacity for T cell function, and DC homing to draining lymph node [30] (Table 1). Tumor hypoxia can further complicate the immune response by modulating expression of various molecules in cancer cells that are necessary for developing proper anti-tumor immunity. For example, tumor hypoxia has been reported to increase programmed death-ligand 1 (PD-L1) expression via activating hypoxia-inducible factor-1 (HIF-1) transcription factor in clear cell renal cell carcinoma [31]. PD-1 is an immune checkpoint receptor on T cell whose ligands, PD-L1 and PD-L2, are commonly being expressed in cancer cells and antigen presenting cells such as dendritic cells and macrophages [323334]. Engage of PD ligands to PD-1 leads to down-regulation of immune responses by blocking ZAP-70 phosphorylation and association with CD3-ζ [3536]. This signaling results in reducing PKC-θ activation, which activates NF-κB transcription factor leading to production of pro-inflammatory cytokines such as IL-2 [37]. Hence the use of PD-1 antibodies such as nivolumab, pembrolizumab, and pidilizumab, or PD-L1 antibodies including PD-L1BMS-936559, MPDL3280A, and MEDI-4736 are receiving much attention with a hope of much improved anti-tumor responses [38]. Hypoxia has also shown to decrease proliferation of CD8+ tumor-infiltrating lymphocytes (TILs) and induce IL-10 immunosuppressive cytokine production [39].
Abscopal effect, originally proposed by R.H. Mole in 1953, describing the effect of radiation to the distant tumor site after local irradiation within the same organism [40], is probably the ultimate proof that irradiation can trigger potent anti-tumor response systemically. It has been proposed that this is mediated by radiation-induced anti-tumor T cells [41]. A number of preclinical studies have demonstrated that addition of other strategies including Flt3 ligand [42], macrophage inflammatory protein-1α (MIP-1α) [43], or vaccine against tumor-associated antigen such as carcinoembryonic antigen (CEA) [44] can lead to tumor-specific abscopal effects by irradiation in mice. However, the fact that abscopal effect has been reported to be very rare both preclinically and clinically [20] and that the exact radiotherapy regimen responsible for such effect are not yet known suggest that much further work is needed to establish radiation-induced anti-tumor immunity.
Pro-tumor immune responses
It has been extensively reported that irradiated cancer cells or tumors including stroma produce cytokine(s) and chemokine(s) such as tumor necrosis factor-α (TNF-α), IL-1α, IL-1β, IL-6, GM-CSF, and transforming growth factor-β(TGF-β) [204546]. Released chemokines including C-C motif chemokine ligand 2 (CCL-2) and C-X-C motif chemokine ligand 12 (CXCL-12) can then act potently to recruit TILs into irradiated tumors [47]. There are many mechanisms by which ionizing radiation promotes the release of chemokines from tumors. For example, irradiation can result in upregulation of HIF either by killing aerobic cells resulting in an increase in tumor hypoxia [4849] or by reactive oxygen species (ROS) [50]-mediated inhibition of proline hydroxylase [51], the enzyme responsible for degrading HIF-α subunits [52]. The HIF transcription factor is known to be able to induce numerous cytokines, chemokines, and growth factors including CXCL-12, CCL-2 and vascular endothelial growth factor (VEGF) [535455] (Fig. 1), all of which can potently recruit TILs. The TILs express cognate receptors for many of these chemokines, for example C-X-C motif chemokine receptor-4 (CXCR-4) for CXCL-12; and C-C motif chemokine receptor-7 (CCR7) for CCL-19 or CCL-21; vascular endothelial growth factor receptor-1 (VEGFR-1) for VEGF and these interactions may further amplify the immune response. Although T-lymphocytes are also known to express CXCR-4, which in theory should be able to be recruited to CXCL-12-expressing tumors and potentiate 'anti-tumor immunity', a number of studies have shown that it is mostly myeloid cells including monocytes and macrophages that largely express CXCR-4 thereby being attracted to CXCL-12-expressing cancer cells [5657] (Fig. 1). Recruited monocytes can then reconstruct the irradiated and thereby being damaged tumor vasculature by expressing matrix metalloproteinase-9 (MMP-9) [58], S100 calciumbinding protein A8 (S100A8) chemoattractant proteins [12], or by releasing VEGF by themselves [59]. Other mechanisms by which irradiation can promote cytokine/chemokine secretion also include activation of NF-κB pathways, which results in production of various pro-inflammatory cytokines/chemokines including TNF-α, IL-1, and CXCL-12, which could then recruit TILs and induce pro-inflammatory microenvironment [60].
Recruited TILs can then further release, even higher concentrations of many of the cytokines listed above or other cytokines, such as IL-1β, IL-6, IL-10, TNF-α, and TGF-β [20]. It has been reported that CD4+ T cells are a major source for TGF-β production [61626364] and that TGF-β regulates activation of CD8+ T cells and natural killer T (NKT) cells, maintenance of peripheral Foxp3-expressing regulatory T cells (Tregs), and survival of CD4+ T cells [65]. Although TGF-β may elicit and potentiate anti-tumor CD8+ T cells, Tregs may counteract such anti-tumor activity by exerting immune suppression in co-operation with myeloid-derived suppressor cells (MDSCs) and tumor-associated macrophages (TAMs).
MDSCs and TAMs may interfere with CD8+ T cell functions in various ways. For examples, it has been shown that they express high levels of arginase-1 (Arg-1) thereby lowering arginine pool for T cell activation and responses [66]; they can also sequester cysteine thereby limiting the availability of cysteine, an amino acid essential for T cell proliferation [50]; they may destroy T cell receptors (TCRs) by producing various ROS [67].
Tumor hypoxia may play an additional role in potentiating these pro-tumor immune responses. Indeed, it has been reported that TAMs are more likely to be polarized towards M2-like pro-tumor phenotype by tumor hypoxia via activation of HIF transcription factor [6869] and that HIF can modify TAM functions such that it increases activities of Arg-1 and NADPH oxidase, which can then further compromise CD8+ cytotoxic functions towards cancer cells [50]. Both tumor hypoxia and radiation has been shown to induce epithelial mesenchymal transition (EMT) of certain cancers [707172]. EMT is a key developmental program often activated during cancer invasion and metastasis. It is currently highly controversial whether EMT triggers anti-tumor response or provokes immune evasion. Although it is possible that anti-tumor response may develop against EMT, it has been reported that SNAIL and ZEB families, transcription factors key to EMT process, are associated with increased CD4+/Foxp3+ Tregs and impaired dendritic cell functions in melanomas [73] and modulate PD-L1 in lung cancer cells [74]. Furthermore, TAMs have been reported to facilitate EMT in pancreatic cancer cells by IL-10 signaling pathway and other molecular mechanisms including increased matrix metalloproteases activities [75].
FACTORS TO CONSIDER UPON COMBINING RADIOTHERAPY WITH IMMUNOTHERAPY
Tumor hypoxia
We outlined above how hypoxia may negatively impact the effect of anti-tumor immunity towards cancers. Therefore, it would be essential to understand how tumor hypoxia changes along the course of radiotherapy. It is generally believed that ionizing radiation of solid tumors would initially result in an increase in hypoxic fractions. This is because DNA radicals produced by ionizing radiation can only be permanently fixed to give rise the DNA damage that can lead to cell death only in the presence of the molecular oxygen, O2 [2]. Thus, radiation-induced cell kill would be initially confined to those of well-oxygenated cancer cells, leaving hypoxic tumor cells viable. Despite such obvious expectation, a number of recent studies indicate that this is not the case. It has been shown by using 18F-misonidazole (F-MISO) radioactive tracer and positron emission tomography (PET) to monitor the dynamic changes in intratumoral hypoxia that 10 and 20 Gy ablative dose irradiated human head and neck squamous cell carcinoma xenografts had minimal changes in the intratumoral hypoxia [76]. Our recent work with F-MISO has also demonstrated that there is no immediate increase in tumor hypoxia following 15 Gy ablative radiation [77]. Although the degree of tumor hypoxia may not change dramatically after irradiation, the extent to which each individual may have in their tumors could be quite different. More importantly, tumor hypoxia is a dynamic process in which it constantly changes spatiotemporally [78], which would affect the local immune cell functions thereby the immune responses.
Radiotherapy regimen
Radiotherapy regimen would also influence the immune responses. Although ionizing radiation can increase MHC class I expression in a dose-dependent manner from 1 Gy up to 25 Gy and that the response could be maintained for up to 3 days [79], the anti-tumor immunity established by tumor-reactive T cells has been reported to be offset at the highest dose by an increase in Tregs [25]. Although preclinical study has reported that fractionated regimen of 7.5 Gy/fraction as optimal for induction of anti-tumor immunity [25], we have recently demonstrated that fractionated irradiation can actually deplete tumor-infiltrating T cells leading to the tumor and metastasis recurrences [80]. In a preclinical study by Lugade and colleagues [81] it was shown that a single high dose irradiation of 15 Gy is superior to fractionated (5×3 Gy) irradiation in inducing anti-tumor activities such as increased levels of antigen presenting cells and interferon-γ(IFN-γ) production in the lymph nodes. However, much further work needs to be done to investigate the detailed molecular mechanisms and immune responses associated with radiotherapy.
CONCLUSIONS
It is an exciting time to have a superb technical advancement in radiotherapy delivering ablative radiation doses precisely to the tumor bearing volume. Furthermore, promising clinical results of checkpoint blockades/immunotherapy further boost the initiative of combining radiotherapy with immunotherapy. In theory, this advanced technique of radiotherapy can significantly boost anti-tumor immune responses. In this review, we have outlined how tumor hypoxia, a major limiting factor contributing failures of chemotherapy and radiotherapy, can further complicate the anti-tumor immune responses. Because tumor hypoxia is a dynamic pathophysiological feature in many solid tumors, it will be essential to investigate the real-time changes in tumor hypoxia with radiotherapy and how immune cells respond towards this system. We believe this topic is of interest to not only cancer biologists, oncologists, and radiation oncologists but also numerous immunologists, which will soon bring us exciting imaging tools for monitoring dynamics of tumor hypoxia and real-time imaging of various populations of immune cells simultaneously.

 XML Download
XML Download