

 PDF
PDF ePub
ePub Citation
Citation Print
Print


INTRODUCTION
The proteasome inhibitor, bortezomib, directly inhibits proliferation and induces apoptosis of multiple myeloma (MM) cells, inhibits mitogen-activated protein kinase-mediated growth signaling in myeloma cells, overcomes drug resistance, enhances the anti-myeloma activity of dexamethasone, and overcomes the resistance to apoptosis of myeloma cells exposed to interleukin-6 (IL-6) [1]. Bortezomib also inhibits the paracrine growth of MM cells by decreasing both cellular adherence to bone marrow (BM) stromal cells (BMSCs) and the associated nuclear factor kappa B-dependent induction of IL-6 secretion by the BMSCs [12]. In parallel, bortezomib exhibits marked clinical activity when administered as a monotherapy to treat even relapsed, refractory MM [3]. Additionally, sub-toxic concentrations of bortezomib potently sensitized myeloma cell lines and patients' cells to DNA-damaging chemotherapeutic agents, including doxorubicin and melphalan. Such cells included those resistant to the drugs and cells isolated from a patient who had relapsed after bortezomib monotherapy [2]. These findings provide a rationale for the use of combination therapy to treat MM.
Progression of MM is dependent upon cellular interactions within the BM microenvironment. Many studies have shown that BMSCs express paracrine factors, including cytokines, and engage in cell-cell contact, both of which play roles in the growth and survival of MM cells and the progression of osteolytic bone disease. Adhesion molecules on the myeloma cell surface mediate the localization of such cells in the BM via binding of the cells to extracellular matrix proteins and BMSCs [45]. Contact between BMSCs and tumor cells, and the secretion of transforming growth factor by the latter, trigger IL-6 secretion from BMSCs and paracrine tumor cell growth [67]. In addition, BMSCs play a role in the adhesion-mediated drug resistance of MM [8910]. Unlike normal mesenchymal stem cells (MSCs), myeloma MSCs express increased basal levels of interleukin-1 (IL-1) beta and tumor necrosis factor (TNF) alpha [11], which may be biologically relevant for the growth and survival of MM cells. Additionally, myeloma BMSCs have been shown to create a microenvironment supporting the growth of MM stem cells [12].
Thalidomide, and immunomodulatory analogs thereof, not only directly induce apoptosis or growth arrest of myeloma cells but also alter the adhesion of myeloma cells to BMSCs, inhibit production of IL-6 and vascular endothelial growth factor (VEGF) in the BM, and stimulate natural killer cell anti-myeloma immunity [13]. Thalidomide also slightly increases the expression levels of IL-1β and TNF-α in BMSCs [14]. Treatment of MSCs with lenalidomide does not affect their growth rate, proliferation, osteogenic, or adipogenic differentiation potential, or their capacity to inhibit T-cell proliferation. However, lenalidomide modulates the expression of MSC cell surface molecules and chemokine secretion in vitro [15], which may contribute to the clinical effects produced by the compound when administered to MM patients. These observations suggest that the therapeutic effects of some anti-myeloma agents are exerted, at least in part, via modulation of BMSC activities.
Bortezomib has been reported to affect both myeloma cells and the BM microenvironment. For example, bortezomib triggered dose-dependent inhibition of VEGF and IL-6 secretion by MM patient-derived endothelial cells [16]. In addition, bortezomib induced interferon (IFN)-γ expression in BMSCs [17]. However, the influence of bortezomib on BMSC biology has not been fully elucidated. In the present study, we examined the roles played by bortezomib in terms of the survival and growth of BMSCs in vitro.
Go to : 
MATERIALS AND METHODS
Cells and reagents
Murine BM stromal MS-5 cells were grown in minimal essential medium-α (MEM-α; Gibco-BRL Life Technologies, Grand Island, NY) supplemented with 10% (v/v) fetal bovine serum (FBS); cells were passaged weekly. MS-5 cells support the proliferation of primitive human hematopoietic cells in long-term culture, and these cells secrete chemokine (CXC motif) ligand 12 (CXCL12), also termed stromal cell-derived factor-1 (SDF-1) [18]. After informed consent was obtained, fresh BM samples were obtained from MM patients at the time of diagnosis, and from healthy donors, for allogeneic hematopoietic stem cell transplantation. BMSCs were prepared by culturing BM mononuclear cells in Isocove's modified Dulbecco's medium (IMDM; Gibco) supplemented with 10% (v/v) FBS. After confluence was attained, adherent cells were harvested and maintained in the same growth medium, with weekly passaging. After three passages, neither CD45-nor PECAMA-1-positvie cells were detected by flow cytometry. Human myeloma RPMI8226 and U266 cells were purchased from the American Type Culture Collection (ATCC; Manassas, VA) and cultured in RPMI-1640 medium (Gibco) supplemented with 10% (v/v) FBS. Recombinant human CXCL12α (SDF-1α) was purchased from R&D Systems (Minneapolis, MN). Bortezomib was a gift from Jassen Korea Ltd. (Seoul, Korea). Dexamethasone was purchased from the Sigma Chemical Co. (St. Louis, MO).
Flow cytometry
To detect apoptosis, cells were stained with fluorescein isothiocyanate (FITC)-conjugated annexin V (BD PharMingen, San Diego, CA) and phycoerythrin (PE)-conjugated propidium iodide (PI; BD PharMingen) at 4℃ for 30 min, and analyzed by flow cytometry using either a Coulter Elite (Coulter Electronics Ltd., Hialeah, FL) or a FACS Canto II (BD Pharmingen) instrument. For cell cycle analysis, cells were stained with PI.
Reverse transcription-polymerase chain reaction (RT-PCR)
Total RNA was prepared from cells using the TRIzol reagent (Gibco), according to the manufacturer's instructions. After purification, 1-µg amounts of RNA were reverse-transcribed using SuperScript reverse transcriptase (Gibco) and the universal primer oligo (dT)15 (Promega, Madison, WI). In each reaction, 1 µL of cDNA was added to 24 µL of PCR buffer (Gibco) supplemented with 2 mM MgCl2, 0.2 µM of each primer, and 1 U Koma Taq polymerase (Koma International, Seoul, Korea). Using a GeneAmp PCR system (Perkin Elmer, Norwalk, CT), 30 cycles of 1 min at 94℃, 45 sec at 55-65℃, and 1 min at 72℃, were performed. The following primers were used: human CXCL12 (sense, AGA ATT CAT GAA CGC CAA GG; anti-sense, AGG ATC CTC ACA TCT TGA ACC); and human glyceraldehyde 3-phosphate dehydrogenase (GAPDH) (sense, CAT GTG GGC CAT GAG GTC CAC CAC; antisense, TGA AGG TCG GAG TCA ACG GAT TTG GTC).
Real-time quantitative RT-PCR (RQ-PCR)
Quantification of mRNAs encoding human CXCL12 and GAPDH was achieved by two-step RQ-PCR performed using a Rotor-Gene 6000 thermal cycler (Corbett Research, Mortlake, Victoria, Australia) and the SyBR Green PCR Master Mix reagent (Qiagen, Hilden, Germany). The amplification conditions were as follows: 15 min at 95℃, 50 cycles of 95℃ for 10 sec, 50-65℃ for 15 sec, and 72℃ for 20 sec.
Migration assay
Cells (2×105/well) were loaded into the upper chambers of 24-well Transwell plates fitted with 5-µm pore diameter membranes (Corning-Costar, Cambridge, MA), and allowed to migrate into the lower chambers for 4 hr. Cells that migrated were counted, and the fold increases in the numbers of migrated cells (compared to that of the control sample) were calculated to yield migration index values. To explore the migration and localization of myeloma cells to positions underneath BMSCs, MS-5 monolayers were treated with bortezomib, washed, and then overlaid with RPMI8226 cells. The localization of RPMI8226 cells under BMSCs was explored using inverted microscopy after 24 hr.
Cell proliferation assay
The effects of bortezomib on BMSC proliferation were evaluated using a colorimetric assay kit (CCK-8 assay kit; Dojindo Laboratories, Tokyo, Japan), based on the MTT assay, according to the manufacturer's instructions. Briefly, 5×103 MS-5 cells or primary BMSCs were incubated in 96-well plates in serum-free X-VIVO medium (BioWhittaker, Walkersville, MA). After incubation of cells with or without bortezomib (5-500 nM) for up to 72 hr, 10 µL CCK-8 solution (provided by the manufacturer) were added to each well. The optical density (OD) was measured 2 hr later using a spectrophotometer (Molecular Devices Co., Sunnyvale, CA) and the -fold-increases in OD values compared to that of the control at commencement of incubation were calculated to yield relative proliferation indices.
Western blot analysis
Cells were starved in serum-free medium for 12 hr, collected by centrifugation, washed in phosphate-buffered saline, and lysed in sodium dodecyl sulfate (SDS) sample buffer (187.5 mM Tris-HCl [pH 6.8], 6% [w/v] SDS, 10% glycerol, 150 mM DTT, and 0.03% [w/v] bromophenol blue). Equal amounts of protein from each sample were separated by electrophoresis on 10% (w/v) SDS-polyacrylamide gels and transferred to polyvinylidene fluoride membranes (Amersham Life Science, Arlington Heights, IL). The membranes were blocked for 1 hr in Tris-buffered saline (TBS) containing 5% (w/v) milk and 0.1% (v/v) Tween 20, and incubated with a primary mouse monoclonal antibody raised against CXCL12 (Cell Signaling Technology Inc., Danvers, MA) overnight at 4℃. The blots were washed with TBS containing Tween 20, incubated with goat anti-mouse CXCL12 polyclonal antibody (Thermo Scientific, Barrington, IL) for 2 hr, and developed using West-Zol Plus (iNtRON Biotechnology, Seoul, Korea).
Enzyme-linked immunosorbent assay (ELISA)
The levels of CXCL12α secreted into media by cultured cells were measured using commercial ELISA kits (R&D Systems) according to the manufacturer's instructions. Cells (5×105) were incubated in 1 mL serum-free X-VIVO medium in 24-well plates at 37℃ under 5% (v/v) CO2. After 72-hr incubation, culture supernatants were harvested and analyzed by ELISA. OD values were measured using a spectrophotometer (Molecular Devices Co.). Standard curves were constructed and levels of secreted CXCL12α calculated using the Softmax software (Molecular Devices Co.). The serum levels of CXCL12α were measured in an identical manner.
Knockdown of CXCL12 using siRNA
Cells were seeded into 12-well plates (3×105/well), incubated for 5-10 min at room temperature, and then transfected with 5-25 nM CXCL12 siRNA or control siRNA (Qiagen) using the HiperFect transfection reagent (Qiagen), according to the manufacturer's instructions. Cells were cultured in normal growth medium for 24 hr after transfection. The transfection efficiency of control siRNA was evaluated by fluorescence microscopy. The extent of suppression of CXCL12 synthesis was determined by RQ-PCR and ELISA.
Statistical analyses
Results are expressed as the mean and standard deviation (SD) of data obtained from at least three experiments. Data were analyzed using Student's t-test for the comparison of paired samples. Statistical data were obtained using the SPSS ver. 17.0 (SPSS Inc., Chicago, IL). A P value <0.05 was deemed to indicate statistical significance.
Go to :
RESULTS
Bortezomib inhibits the proliferation of BMSCs
Bortezomib inhibited the spontaneous proliferation of BMSCs in serum-free X-VIVO medium, in a dose-dependent manner. Incubation of MS-5 cells with 5 nM bortezomib for up to 3 days significantly decreased cell proliferation, as compared with the control cell value (relative proliferation indices: 2.1±0.5 vs. 1.6±0.3 at 48 h; and 3.5±0.6 vs. 1.5±0.2 at 72 h; both P values <0.05), and ≥50 nM bortezomib abolished cell proliferation. Similar results were obtained using BMSCs from 3 healthy individuals and 5 myeloma patients. Consistent with these findings, bortezomib reduced the proportions of MS-5 cells in S phase in a concentration-dependent manner (Fig. 1).
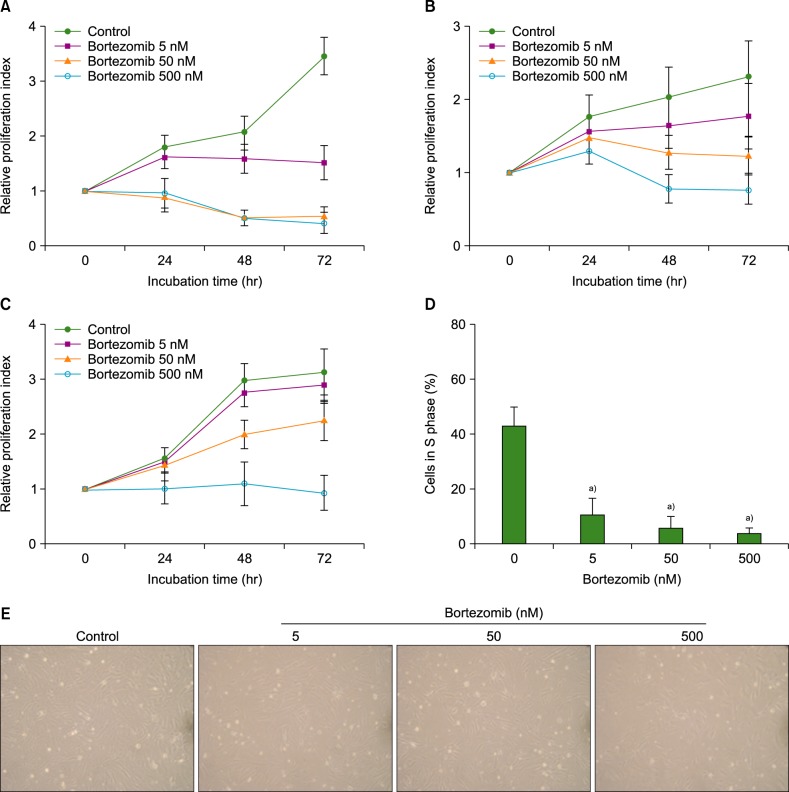 | Fig. 1Bortezomib inhibits the proliferation of bone marrow stromal cells (BMSCs). MS-5 cells (A), BMSCs from 3 healthy individuals (B), and BMSCs from 5 myeloma patients (C) were incubated without or with bortezomib (5-500 nM) in 96-well plates in serum-free X-VIVO medium, and cell proliferation was measured by colorimetric assay. Data are the mean±SD of the relative proliferation indices from 3 independent experiments. (D) MS-5 cells were incubated with or without bortezomib for 24 hr prior to cell cycle analysis. a)P<0.05, as compared with the control (no bortezomib). (E) BMSCs from a normal individual, incubated for 24 hr, are shown.
|
Bortezomib induces delayed apoptosis of BMSCs
Bortezomib, even at lower concentrations, rapidly and markedly induced apoptosis of U266 myeloma cells. Thus, only 10% of U266 cells remained alive after 24-hr incubation with 5 nM bortezomib. In contrast, bortezomib (5-500 nM) did not affect the survival of MS-5 cells after 24-hr incubation. However, 50 nM and 500 nM bortezomib markedly increased the proportions of annexin V-positive apoptotic cells after 72 hr, as compared with the control values (0.9±0.2% vs. 80.1±5.6% for 50 nM bortezomib; 0.9±0.2% vs. 82.7±6.8% for 500 nM bortezomib; both P values <0.05). Similar results were obtained when BMSCs from 3 healthy individuals and 5 myeloma patients were tested (Fig. 2).
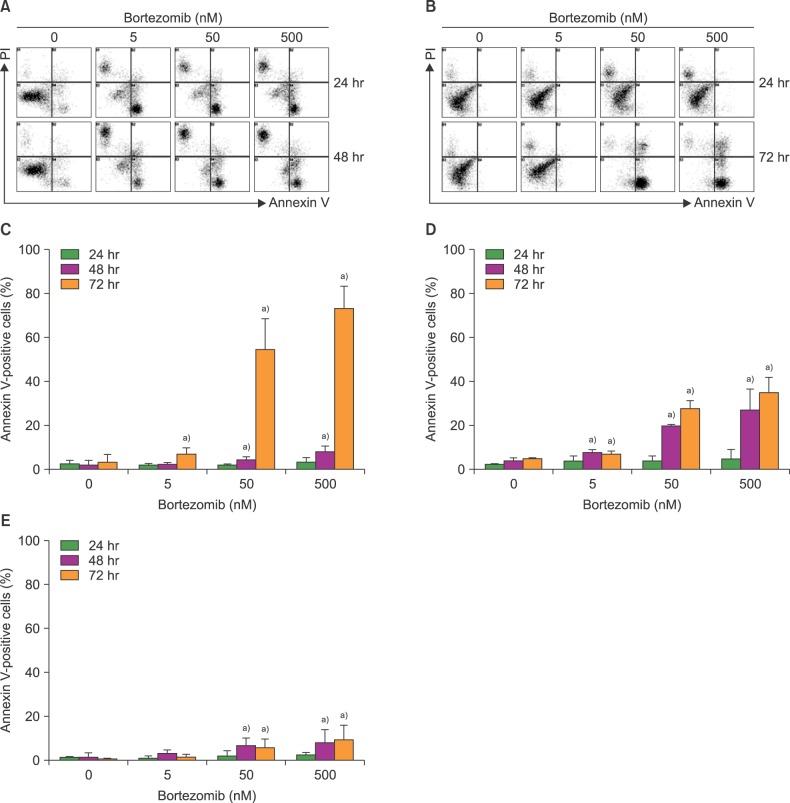 | Fig. 2Bortezomib induces delayed apoptosis of bone marrow stromal cells (BMSCs). Cells were incubated in appropriate growth media, without or with bortezomib (5-500 nM), for 24-72 hr. Apoptosis was measured by flow cytometry after staining the cells for annexin V. (A) Representative flow cytometric profiles of bortezomib-treated U266 myeloma cells. (B) Representative flow cytometric profiles of bortezomib-treated MS-5 cells. (C) Apoptosis of MS-5 cells induced by bortezomib. Data are the mean±SD of 3 independent experiments. (D) Apoptosis of bortezomib-exposed BMSCs from 3 healthy individuals. Data are the mean±SD. (E) Apoptosis of bortezomib-exposed BMSCs from 5 myeloma patients. Data are the mean±SD. a)P<0.05, as compared with the control (no bortezomib).
|
CXCL12 is an autocrine growth factor for BMSCs
We hypothesized that downregulation of the chemokine, CXCL12, might be involved in bortezomib-induced inhibition of BMSC survival and proliferation. As a first step toward testing this hypothesis, we examined the spontaneous proliferation of BMSCs with siRNA-mediated knockdown of CXCL12 mRNA production. Knockdown of CXCL12 mRNA in BMSCs from both healthy individuals and myeloma patients significantly decreased the spontaneous proliferation of such cells (Fig. 3), and addition of CXCL12α partially restored proliferation (data not shown), indicating that CLCX12 acted as an autocrine growth factor for BMSCs.
 | Fig. 3Knockdown of chemokine (CXC motif) ligand 12 (CXCL12) inhibits the spontaneous proliferation of bone marrow stromal cells (BMSCs). BMSCs from 3 normal individuals (A) and 3 multiple myeloma patients (B) were transfected with 25 nM CXCL12 siRNA or control siRNA for 24 hr, incubated for up to 72 hr, and subjected to cell proliferation assays. a)P<0.05, compared with the control siRNA.
|
Bortezomib downregulates CXCL12 expression and production in BMSCs
Next, we examined whether bortezomib affected the expression of CXCL12 mRNA or protein in BMSCs. Treatment of MS-5 cells with bortezomib at 5, 50, and 500 nM for 24 hr reduced the levels of CXCL12 mRNA expression to 50%, 20%, and <10% that of the control, respectively. In parallel, bortezomib decreased CXCL12 production in a concentration-dependent manner (Fig. 4). Similar results were obtained when BMSCs from 3 healthy individuals and 5 multiple myeloma patients were tested (Fig. 5A-D). The serum levels of CXCL12α in myeloma patients (N=3) were significantly reduced after 3 days after one intravenous administration of bortezomib at 1.3 mg/m2 (453±124 pg/mL CXCL12α vs. 145±87 pg/mL CXCL12α; P <0.05) (Fig. 5E). Media conditioned by MS-5 cells treated with 5 nM bortezomib induced less chemotaxis of RPMI8226 myeloma cells than did media conditioned by non-treated cells (migration indices: 7.3±1.5 vs. 3.4±1.1; P <0.05) (Fig. 5F).
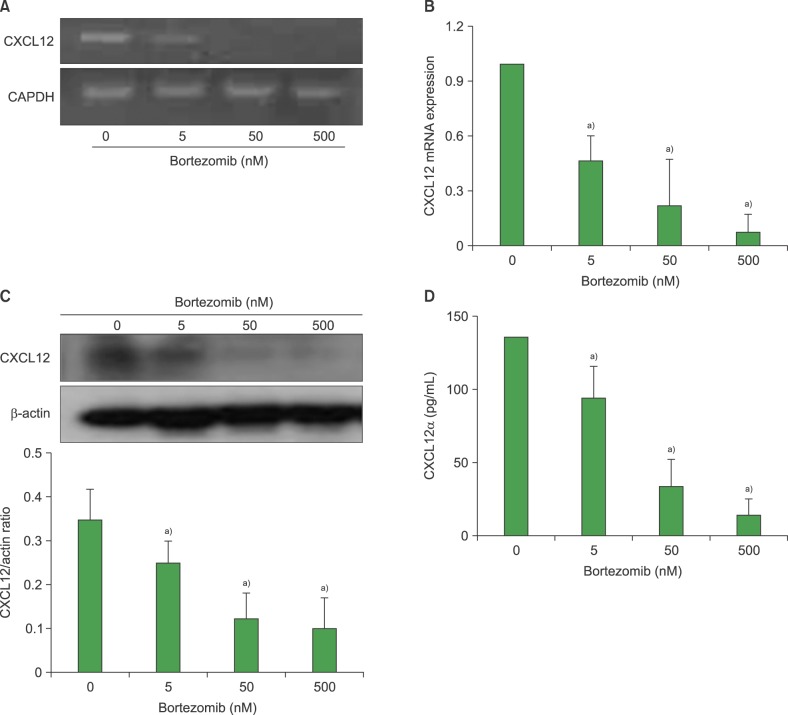 | Fig. 4Bortezomib downregulates the expression and production of chemokine (CXC motif) ligand 12 (CXCL12) in MS-5 cells. MS-5 cells were incubated without or with bortezomib (5-500 nM) in X-VIVO medium for 24 hr prior to reverse transcription-PCR (A), real-time quantitative reverse transcription-PCR (B), and western blotting for CXCL12 (C). GAPDH, glyceraldehyde 3-phosphate dehydrogenase. (D) Concentrations of CXCL12α in 3-day MS-5 culture supernatants were measured by ELISA. Data are mean±SD of 3 independent experiments. a)P<0.05, compared with the control (no bortezomib).
|
 | Fig. 5Bortezomib downregulates the expression and production of chemokine (CXC motif) ligand 12 (CXCL12) in bone marrow stromal cells (BMSCs). BMSCs from 3 normal individuals (A, B) and 3 multiple myeloma patients (C, D) were incubated without or with bortezomib (5-500 nM) in serum-free X-VIVO medium. After 24 hr, CXCL12 mRNA levels were measured using quantitative reverse transcription-PCR (A, C). After 72 hr, media concentrations of CXCL12α were measured by ELISA (B, D). a)P<0.05, compared with the control (no bortezomib). (E) Serum concentrations of CXCL12α before, and 3 days after, intravenous administration of bortezomib (1.3 mg/m2) to multiple myeloma patients (N=3). a)P<0.05. (F) Transmigration of RPMI8226 cells induced by 3-day bortezomib-treated or -non-treated MS-5 cell culture media. a)P<0.05, compared with the control (no bortezomib). (G) MS-5 cell monolayers were treated with (right panel) or without (left panel) 5 nM bortezomib for 24 hr. RPMI8226 cells were added to the MS-5 monolayers, and migration of cells beneath the monolayers (the dark phases) was studied by inverted microscopy 24 hr later. Short-term bortezomib treatment inhibited the localization of RPMI8226 cells under monolayers. Representative results are shown.
|
Bortezomib inhibits the localization of myeloma cells under BMSC monolayers
To examine the migration of myeloma cells beneath stromal cells, RPMI8226 cells were added to MS-5 feeder layers in X-VIVO medium. After 24 hr, many of the former cells had migrated beneath the feeder layers, as was readily apparent using inverted microscopy. Short-term bortezomib treatment of MS-5 monolayers reduced the extent of RPMI8226 cell localization under the monolayers, as compared with non-treated MS-5 cells (Fig. 5G), indicating that bortezomib-induced alterations in BMSCs that affected myeloma cell biology.
Bortezomib and dexamethasone exert additive effects in downregulating BMSC CXCL12 production
We explored whether bortezomib interacted with corticosteroids to modulate CXCL12 production by BMSCs. Dexamethasone (0.1-10 µM) downregulated CXCL12 production in a concentration-dependent manner, as evidenced by western blotting and ELISA analyses. RQ-PCR and ELISA of CXCL12 levels revealed that bortezomib and dexamethasone exerted additive effects on the downregulation of CXCL12 mRNA and protein levels (Fig. 6).
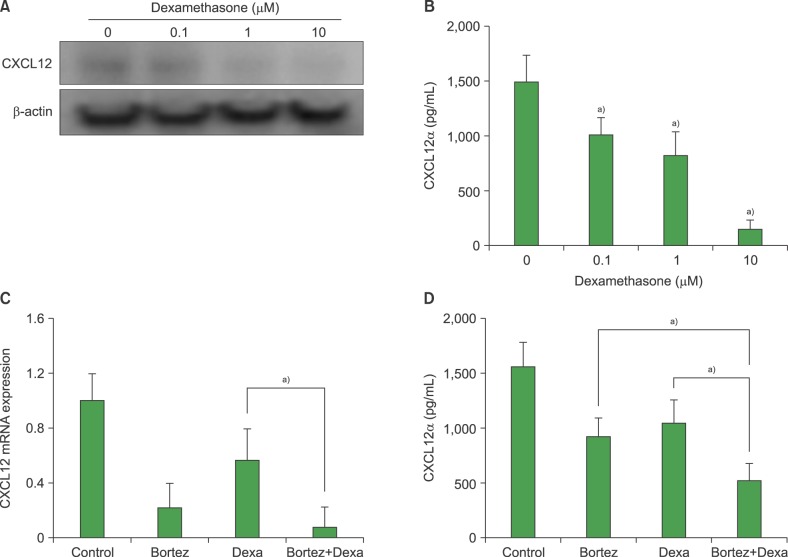 | Fig. 6Bortezomib and dexamethasone exert an additive downregulation of chemokine (CXC motif) ligand 12 (CXCL12) levels in MS-5 cells. (A) MS-5 cells were treated with dexamethasone for 24 hr prior to western blotting for CXCL12. (B) Concentrations of CXCL12α in 3-day MS-5 culture media were measured by ELISA. Data are the mean±SD of 3 independent experiments. a)P<0.05, compared with the control (no bortezomib). (C) MS-5 cells were treated with bortezomib (5 nM) and/or dexamethasone (Dexa; 0.1 µM) and CXCL12 mRNA was measured by quantitative reverse transcription-PCR. (D) MS-5 cells were incubated with bortezomib (5 nM) and/or dexamethasone (0.1 µM) in X-VIVO medium for 72 hr. Media concentrations of CXCL12α were measured by ELISA. a)P<0.05.
|
Go to :
DISCUSSION
In the present study, we showed that bortezomib inhibited the proliferation of BMSCs in a concentration-dependent manner in vitro, irrespective of MM status. Not only pharmacological concentrations of bortezomib (≥50 nM), but also 5 nM bortezomib (the approximate 50% inhibitory concentration [IC50] for myeloma cell lines), significantly inhibited BMSC proliferation. These results suggest that bortezomib can affect BMSCs under both normal and pathological circumstances. Bortezomib induced apoptosis of BMSCs obtained from both healthy individuals and MM patients, in a concentration-dependent manner. Interestingly, apoptosis of BMSCs became evident after incubation for ≥48 hr. It is well known that bortezomib directly induces marked apoptosis of myeloma cells after several hours [1]. Thus, this delay of bortezomib-induced BMSC apoptosis raises the possibility that indirect pathways are involved in this process.
CXCL12 supports the survival and/or proliferation of many cell types, in either a paracrine and/or autocrine manner. For example, trophoblasts produce CXCL12, and exogenous CXCL12 stimulates trophoblast cell proliferation; overexpression of CXCL12 and the receptor CXCR4 induces autocrine/paracrine cell proliferation in human pituitary adenoma tissue [19]; autocrine CXCL12 signaling drives the evolution of tumor-promoting mammary stromal myofibroblasts [20]; gallbladder carcinoma cells produce CXCL12, and the overexpression of CXCL12 enhances cell survival and proliferation [21]. In our previous study, we showed that pertussis toxin inhibited the in vitro proliferation of BMSCs from healthy individuals and in two lines of CXCL12-producing fibroblasts, thus suggesting that CXCL12 served as an autocrine growth factor for BMSCs [22]. Indeed, CXCL12 has been shown to be an autocrine growth factor for selected BM mesenchymal stem cells [23]. In the present study, we showed that siRNA-mediated knockdown of CXCL12 significantly inhibited the spontaneous proliferation and survival of BMSCs derived from both healthy individuals and MM patients. Together, the data clearly suggest that CXCL12 serves as an autocrine growth factor for BMSCs, both in healthy individuals and MM patients.
We showed that bortezomib downregulated the expression and production of CXCL12 in BMSCs from both healthy individuals and MM patients. These results contradict previous observations indicating that both radiation and conventional chemotherapeutic agents induced upregulation of CXCL12, regarded as a compensatory reaction by BMSCs to cellular damage [242526]. As CXCL12 serves as an autocrine factor for BMSCs, it is highly likely that bortezomib inhibits the survival and proliferation of BMSCs at least in part by downregulating CXCL12 expression. We have previously shown that dexamethasone upregulated CXCR4 expression in myeloma cells, while downregulating CXCL12 expression in BMSCs [27]. In the present study, bortezomib and dexamethasone exerted an additive downregulation of CXCL12 in BMSCs. This observation may provide an additional explanation for the enhanced clinical effect noted when a combination of these two agents is used.
In addition to inhibiting the survival and proliferation of BMSCs, bortezomib-induced CXCL12 downregulation affected myeloma cell biology. Conditioned medium from bortezomib-treated MS-5 cells exerted a lower chemotactic activity toward MM cells than the conditioned medium from control cells that had not been exposed to this drug. Additionally, the migration of myeloma cells and the localization of these cells under bortezomib-treated BMSC monolayers significantly reduced, as compared to untreated monolayers, indicating that bortezomib induced segregation of myeloma cells from BMSCs. Given that BMSCs support myeloma cells via both paracrine pathways and cell-cell contact, segregation of myeloma cells from BMSCs would hamper their survival and reduce the growth advantages provided by BMSCs. Indeed, the CXCR4 inhibitor, AMD3100, disrupted the interaction of myeloma cells with the BM microenvironment and enhanced the sensitivity of BMSCs to chemotherapy [28]. Together, these data suggest that bortezomib-induced CXCL12 downregulation not only inhibits BMSC growth, but also eliminates the advantages afforded by cell-cell contact between BMSC and myeloma cells. These effects very likely contribute to the anti-myeloma effects of the drug.
In summary, bortezomib downregulates CXCL12 expression in BMSCs and inhibits the survival and growth of BMSCs, irrespective of MM status. These results suggest that the anti-myeloma effects of bortezomib may be exerted, at least in part, by modulation of BMSC biology.
Go to :
 XML Download
XML Download