

 PDF
PDF ePub
ePub Citation
Citation Print
Print


INTRODUCTION
Many patients acquire cytomegalovirus (CMV) viremia after allogeneic hematopoietic stem cell transplantation (HSCT). This sometimes results in overt CMV disease or fatal outcomes despite pre-emptive treatment. Human CMV is a ubiquitous human herpes virus associated with subclinical primary infections followed by life-long asymptomatic carriage, during which the innate and adaptive immune systems act together to control the virus [1]. However, CMV diseases associated with severe symptoms are easily provoked in immune-suppressed patients like allogeneic HSCT recipients [2]. Immune-suppression can lead to viremia of latent CMV in HSCT recipients, which may progress to overt CMV disease [3]. CMV is closely associated with the immune response as well as with inflammatory factors; CMV viremia is also related to an enhanced secretion of cytokines that can render a patient unable to defend himself against pathogens. This pathway is tightly regulated to prevent excessive inflammatory damage in the host [4, 5].
Suppressors of cytokine signaling (SOCS) proteins are inhibitors of cytokine signaling pathways and key physiological regulators of both innate and adaptive immunity [6]. In addition to regulating the activity of immune cells such as macrophages and dendritic cells, SOCS proteins are also essential for B- and T-cell development and differentiation. The SOCS and cytokine-inducible SRC homology 2 (SH2) protein (CIS) family is comprised of eight members (CIS and SOCS1-SOCS7). All family members have a central SH2 domain, an N-terminal domain of variable length and sequence, and a C-terminal 40-amino acid module called the SOCS box [7]. In particular, the roles of SOCS1 and SOCS3 in toll-like receptor (TLR) responses, which recognize the CMV virus, have been extensively investigated [4, 6].
Recent studies have shown that SOCS1-deficient mice were hypersensitive to lipopolysaccharide (LPS), leading to an increase in tumor necrosis factor-α (TNF-α) and interleukin-12 (IL-12) production. SOCS1 was also demonstrated to be an important inhibitor of both type I and type II interferon (IFN) signaling in vivo [8, 9] and contributes to the balance between the beneficial antiviral and detrimental pro-inflammatory effects of IFN signaling [10]. SOCS3, one of the most abundantly induced proteins in macrophages following stimulation with LPS, is a key regulator of the divergent activities of IL-6 and IL-10 following TLR stimulation [11, 12]. Using a conditional deletion of SOCS3 in mice, Croker et al. [13] showed that SOCS3 negatively regulated IL-6, implying the importance of SOCS3 in IL-6-related immune and inflammatory responses, as well as in pathophysiologic conditions.
Therefore, we investigated the expression of SOCS genes in patients with CMV viremia that received allogeneic HSCT for various hematologic diseases. Our data suggest that expression of SOCS1 and SOCS3 genes in CMV viremia may attenuate a CMV attack by cytokine signaling modulation and may be critical for the prevention and treatment of CMV diseases by coordinating the individual cytokines released in immune-suppressed allogeneic HSCT recipients.
Go to : 
MATERIALS AND METHODS
Human blood sampling and preparation
All experiments were performed with authorization of the Institutional Review Board (IRB) for Human Research at the Catholic University of Korea. Study patients were the recipients of allogeneic SCT that were initially diagnosed with one of the hematologic diseases designated by the World Health Organization (WHO). Heparinized blood samples were collected from the recipients at a time of high CMV DNAemia for those diagnosed with CMV viremia (CMV+ group) and at any time after transplantation for those without CMV viremia (CMV- group). In addition, blood samples were collected from the recipients before conditioning (pre-HSCT group) and healthy donors (healthy donor group) before harvesting hematopoietic stem cells.
Mononuclear cells were isolated by overlaying the heparinized blood samples on a Ficoll-Hypaque gradient (density, 1.077; Lymphoprep; Gibco-BRL, Carlsbad, CA, USA), followed by centrifugation at 400×g for 30 minutes. The buffy coats were harvested and washed twice with phosphate-buffered saline (pH 7.4).
Definitions
We defined patients as positive for CMV viremia if they had a CMV DNA load ≥ 500 copies/mL, which is the lowest detectable level. Acute graft-versus-host disease (aGVHD) was assessed according to previously published criteria [14, 15] and patients with aGVHD grades II-IV were regarded as positive for aGVHD occurrence.
Previously, we demonstrated that SOCS1 and SOCS3 genes behave differently in patients depending on the type and severity of GVHD [16]. Therefore, in this study, we classified the recipients into an additional four subgroups to analyze the expression levels of SOCS genes between recipients with CMV viremia and without CMV viremia, according to the occurrence of aGVHD: a group with both CMV viremia and GVHD (CMV+GVHD+ subgroup), without CMV viremia but positive for GVHD (CMV-GVHD+ subgroup), positive for CMV viremia but without GVHD (CMV+GVHD- subgroup), and the group with neither CMV viremia nor GVHD (CMV-GVHD- subgroup).
CMV prophylaxis, monitoring, and pre-emptive treatment
For the prophylaxis of CMV, acyclovir (10 mg/kg three times a day) was intravenously administered from the conditioning until neutrophil engraftment. Recipients were monitored for CMV DNA load twice a week using quantitative reverse-transcription PCR (qRT-PCR) using a LightCycler 2.0 Real-Time PCR system (Roche Diagnostics, Mannheim, Germany) from neutrophil engraftment to hospital discharge. Thereafter, they were monitored weekly to biweekly until the cessation of immunosuppressive agents. For CMV positive patients, we conducted a risk-adapted pre-emptive therapy to prevent CMV disease according to the treatment protocol of our institution [17].
Real-time quantitative reverse transcription PCR analysis
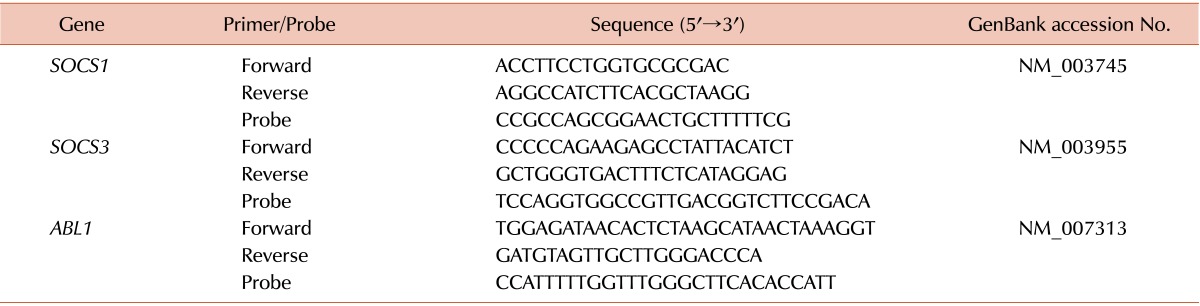
In the present study, we performed qRT-PCR on the blood samples from all recipients and donors as described previously because no data were available regarding the reference levels of SOCS genes [18]. Total RNA was extracted from mononuclear cells using the TRIzol reagent (Invitrogen, Carlsbad, CA, USA). RNA samples were treated with RNase-free recombinant DNase I (Roche, Mannheim, Germany) and subjected to reverse transcription using the Transcriptor First-Strand cDNA Synthesis Kit (Roche). cDNA synthesis was achieved by incubating at 25℃ for 10 minutes and at 42℃ for 60 minutes. Thereafter, the reaction was inactivated by heating at 99℃ for 5 minutes. The qRT-PCR reactions and fluorescence measurements were performed using a LightCycler 480 Real-Time PCR system (Roche). The probe was labeled at its 5' end with 6-carboxy-fluorescein reporter dye and at its 3' end with 6-carboxy-tetramethyl-rhodamine as a sequencer. The qRT-PCR primers and probes for SOCS1, SOCS3, and ABL1 (control genes used for normalization) are listed in Table 1. Quantitative amplification was performed using the following parameters; denaturation at 95℃ for 10 minutes, followed by 50 cycles of denaturation at 95℃ for 10 seconds, annealing and elongation at 60℃ for 30 seconds, and a final cooling step at 40℃ for 30 seconds. All sample analyses were performed in triplicate.
Statistical analysis
All results are presented as the mean ± SE. Statistical analyses were performed with the Mann-Whitney U test for comparisons between 2 groups. P values of P≤0.050 were considered to denote statistical significance. All statistical analyses were performed by GraphPad Prism version 5.0.3 software (GraphPad Software Inc., San Diego, CA, USA).
Go to :
RESULTS
Patient characteristics
Blood samples from 107 recipients with acute myeloid leukemia (N=58), acute lymphoblastic leukemia (N=20), myelodysplastic syndrome (N=12), chronic myelogenous leukemia (N=4), severe aplastic anemia (N=9), and other hematologic diseases (N=4) were collected between 2009 and 2011. Recipients received allogeneic HSCT from HLA-matched siblings (N=51) and unrelated donors (N=56). They received myeloablative conditioning (N=71) and reduced-intensity conditioning (N=36) before allogeneic HSCT. Of them, 61 (57.0%) recipients had CMV viremia at blood sampling. The median CMV DNA load of the CMV+ group was 9,250 copies/mL (range, 675-2,144,711). An additional 17 and 55 blood samples were obtained from the recipients and healthy donors, respectively. Forty recipients (65.8%) with CMV viremia and 29 (63.0%) without CMV viremia were diagnosed with aGVHD. Other baseline and transplant-related clinical characteristics of recipients and donors enrolled in this study are summarized in Table 2.
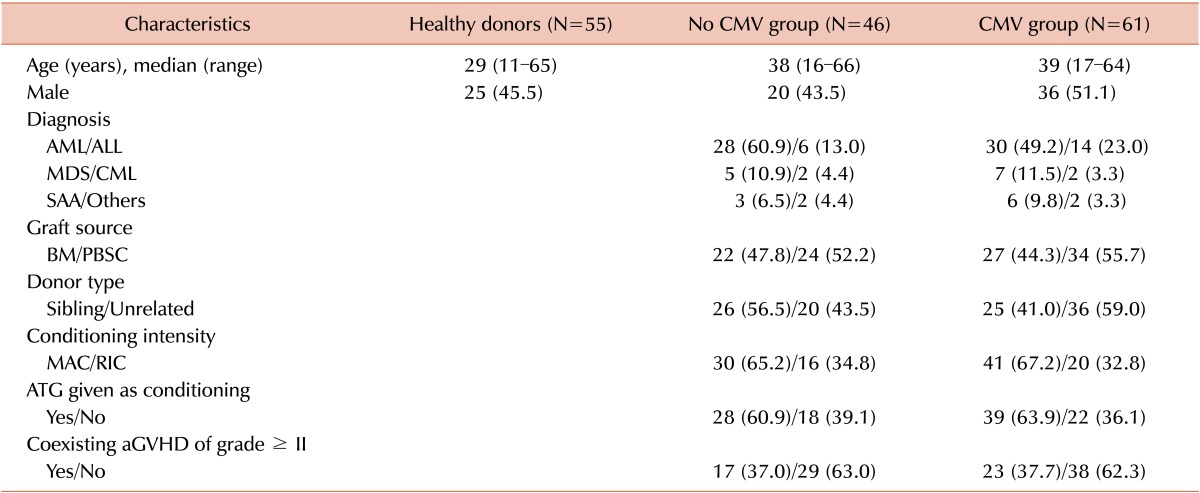
Table 2
Baseline clinical and transplantation-related characteristics of recipients and donors.
Abbreviations: CMV, cytomegalovirus; AML, acute myelogenous leukemia; ALL, acute lymphoblastic leukemia; MDS, myelodysplastic syndrome; CML, chronic myeloid leukemia; SAA, severe aplastic anemia; BM, bone marrow; PBSC, peripheral blood stem cell; MAC, myeloablative conditioning; RIC, reduced intensity conditioning; ATG, anti-thymocyte globulin; aGVHD, acute graft-versus-host disease.
![]()
SOCS1 gene expression in the healthy donor group, pre-HSCT group, and the recipients
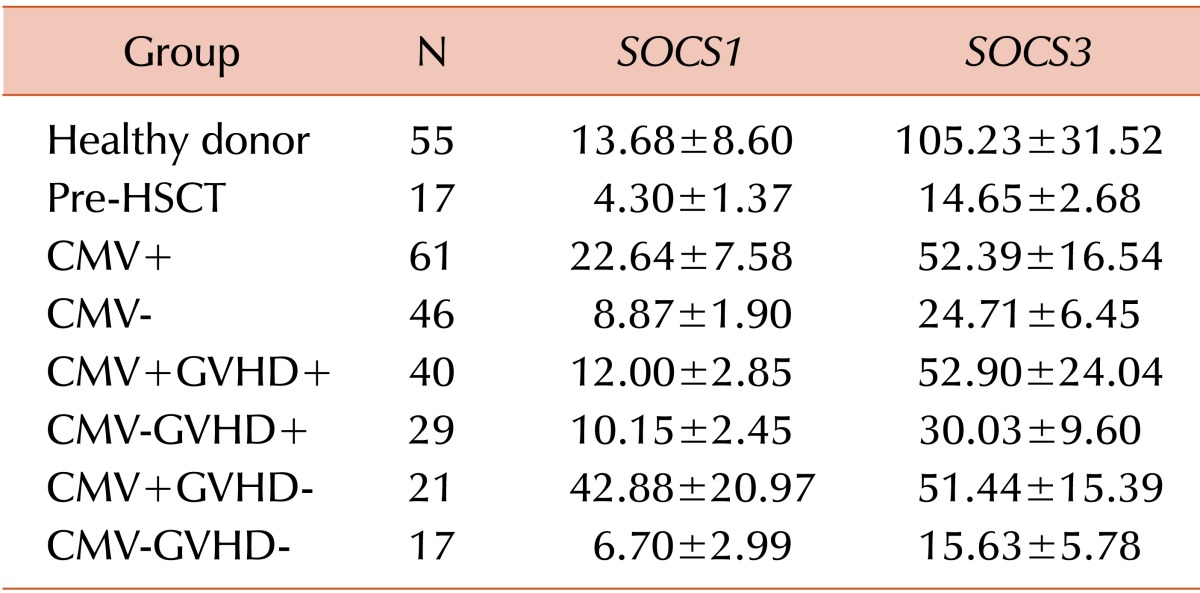
The level of SOCS1 gene expression was not significantly different between the CMV+ group and the CMV- group (22.64 vs. 8.87; P=0.173). When we conducted the pairwise comparison of the SOCS1 gene expression levels of each subgroup, the CMV+ group (22.64 vs. 13.68; P<0.001) and the CMV+GVHD- (42.88 vs. 13.68; P=0.012) subgroup showed significantly higher SOCS1 expression levels than the healthy donor group, whereas the SOCS1 expression levels of the CMV- group (8.87 vs. 13.68; P=0.030), the CMV+GVHD+ subgroup (12.00 vs. 13.68; P<0.001), and the CMV-GVHD+ subgroup (10.15 vs. 13.68; P=0.002) were significantly lower than those of the healthy donor group. In addition, the CMV+ group (22.64 vs. 4.30; P=0.016) and the CMV+GVHD+ subgroup (12.00 vs. 4.30; P=0.014) showed significantly higher SOCS1 expression levels than the pre-HSCT group. However, there were no significant differences between the CMV+GVHD+ subgroup and the CMV-GVHD+ (12.00 vs. 10.15; P=0.666) subgroup and between the CMV+GVHD- subgroup and the CMV-GVHD- subgroup (42.88 vs. 6.70; P=0.144).
SOCS3 gene expression in the healthy donor group, the pre-HSCT group, and the recipients
The SOCS3 gene expression levels were also not significantly different between the CMV+ group and the CMV-group (52.39 vs. 24.71; P=0.125). Subsequently, we conducted a pairwise comparison of SOCS3 expression levels among each subgroup. The SOCS3 expression level of the healthy donor group was significantly higher compared to the pre-HSCT group (105.23 vs. 14.65; P<0.001), CMV+ group (105.23 vs. 52.39; P=0.008), CMV- group (105.23 vs. 24.71; P<0.001), CMV+GVHD+ subgroup (105.23 vs. 52.90; P=0.007), CMV-GVHD+ subgroup (105.23 vs. 30.03; P=0.002), and CMV-GVHD- subgroup (105.23 vs. 15.63; P<0.001). SOCS3 expression in the healthy donor group was not significantly higher than in the CMV+GVHD- subgroup (105.23 vs. 51.44; P=0.155). In addition, the CMV-GVHD- subgroup showed significantly lower SOCS3 expression compared to the CMV+ group (15.63 vs. 52.39; P=0.005), the CMV+GVHD+ subgroup (15.63 vs. 52.90; P=0.010), and the CMV+GVHD- subgroup (15.63 vs. 51.44; P=0.014). However, there was no significant difference between the CMV+ GVHD+ subgroup and the CMV-GVHD+ subgroup (52.90 vs. 30.03; P=0.444). The comparison of SOCS1 and SOCS3 gene expression in the healthy donor group, the pre-HSCT group, and the recipients is shown in Fig. 1. The qRT-PCR experiment data is provided with detailed gene expression levels in Table 3.
 | Fig. 1Comparison of (A) SOCS1 and (B) SOCS3 gene expression levels in the healthy donor group, the pre-HSCT group, and the recipients. Mononuclear cells from recipients were isolated and subjected to real-time quantitative reverse transcription-polymerase chain reaction. Relative gene expression (RE) was normalized to that of ABL1. Error bars indicate±standard error.
|
Go to :
DISCUSSION
Although the results of our study are based on data collected from patients treated using our institution's own approach for various hematologic malignancies, this trial has several limitations: small sample size and restriction on aGVHD. However, as a pilot study in an understudied field, we showed that the SOCS1 and SOCS3 genes might be associated with CMV viremia in allogeneic HSCT recipients. Although it is well known that these genes are involved in some pathophysiologic events in an unbalanced immune system, including GVHD in patients receiving allogeneic HSCT [16, 19], the correlation between the expression of SOCS genes and CMV viremia based on GVHD occurrence has not yet been revealed. Previous studies have also suggested that SOCS1 and SOCS3 may be associated with hematologic malignancies as well as with other diseases [20, 21]. Therefore, it is possible to consider the relevance of CMV viremia with an orchestrated expression of SOCS genes after allogeneic HSCT.
Although many specific prophylactic or pre-emptive anti-viral therapies have been widely used [22], an optimal treatment for CMV diseases remains to be established. In particular, understanding the relationship between the immune network and CMV infection is an emerging challenge to the development of treatment methods for CMV diseases with promising immunotherapies [23, 24]. We herein showed that the SOCS3 expression level was significantly lower in the pre-HSCT group than in the healthy donor group. In addition, SOCS1 gene expression levels were relatively higher in the CMV+ group and the CMV+GVHDsubgroup, whereas those of the CMV+GVHD+ subgroup and the CMV-GVHD+ subgroup were not. We found that SOCS1 gene expression levels were higher in the CMV+GVHDsubgroup than the CMV+GVHD+ subgroup. On the other hand, the SOCS3 gene expression level was significantly lower in the CMV-GVHD- subgroup than in all CMV+ groups, regardless of the development of GVHD. These results suggest that both SOCS1 and SOCS3 play potential roles in CMV viremia after allogeneic HSCT, through different pathways. Recently, CMV infected cells were shown to have impaired levels of signal transducers and activators of transcription (STAT) proteins, although intact phosphorylation was reported. It can abolish intrinsic negative feedback loops by SOCS gene incompetence, which suggests a paradoxical regulation in CMV infection [25].
A previous report showed differential expression of SOCS genes in mice with GVHD [19]. In addition, in a previous human study we found that SOCS genes were relevant in acute and chronic GVHD groups [16]. On the other hand, Cantoni et al. [26] reported a reciprocal relationship between GVHD and CMV viremia. Subsequently, further evaluation found that a regulatory balance of SOCS1 and SOCS3 gene expression in CMV viremia is necessary to elucidate the contribution of cytokine feedback mechanisms in GVHD. Therefore, we further analyzed the expression levels of SOCS1 and SOCS3 between allogeneic HSCT recipients with CMV viremia and those without CMV viremia, according to the occurrence of aGVHD. SOCS1 gene expression was higher in the CMV+ group and the CMV+GVHD- subgroup than in the healthy donor group, whereas it was not increased in the CMV+GVHD+ subgroup. Blalock et al. showed up-regulation of SOCS3 gene expression by STAT3 phosphorylation after murine CMV infection in TH17 cells, suggesting that SOCS3 is expressed after CMV infection [27]. However, we still cannot exclude the possibility that the change in SOCS gene expressions in response to human CMV infection is cell-type-, species-type-, and acute or chronic-status-dependent. Additionally, because steroids are commonly used to treat GVHD, it is possible that the differential expression levels of SOCS genes will not be from CMV viremia but from steroids prescribed for GVHD treatment. Therefore, it is important to understand the interplay of SOCS genes and CMV viremia [28].
The SOCS family of proteins contains eight members, called SOCS1-7 and CIS, which are similar in their structure and function via the Janus kinase (JAK)/STAT pathway [29]. Their negative feedback and signaling cascade functions use many different mechanisms, depending on the cell line studied. Therefore, cytokine induction by SOCS members tends to vary in different conditions [30]. Of note, SOCS molecules may interact with other SOCS members to counter-regulate their function; thus, exploration of this cross-modulation is needed to understand the biology of CMV infection in allogeneic HSCT. Therefore, further investigations should be conducted to reveal the function of other SOCS members, including SOCS4, 5, 6, and 7, in the occurrence of CMV infection and GVHD in allogeneic HSCT. Taken together, our preliminary results provide a new platform for studying the association between CMV immunobiology and SOCS genes.
We showed that SOCS genes were differentially expressed in human CMV viremia with acute GVHD occurrence after allogeneic HSCT, suggesting the possibility of modulation of SOCS genes as a therapeutic target in the future. Therefore, these data may elicit the necessity of further large studies to reveal the exact mechanism of cytokine homeostasis with regard to CMV viremia in similar clinical situations after various organ transplantations.
Go to :
 XML Download
XML Download