

 PDF
PDF ePub
ePub Citation
Citation Print
Print


INTRODUCTION
Hemophilia A (HA) is a common congenital bleeding disorder resulting from a qualitative or quantitative deficiency in coagulation factor VIII (FVIII) [12]. Recently, remarkable progress has been made in understanding the molecular basis of HA. FVIII is a multi-domain protein. It is composed of six domains that are arranged as follows: A1-A2-B-A3-C1-C2 (from N to C terminus) [3]. The FVIII protein is secreted as a heterodimer consisting of a heavy chain (A1-A2-B domains) and a light chain (A3-C1-C2 domains); these bind to each other in circulating blood via Ca2+ and Cu2+ [45]. FVIIIa (the active form of FVIII) functions primarily as a cofactor for coagulation factor IX (FIX) [678]. FVIII also interacts with diverse proteins such as von Willebrand factor (vWF), thrombin, calnexin, and calreticulin [9]. vWF binding occurs when FVIII is secreted into the bloodstream [9]. The FVIII-vWF complex has a longer half-life and is more stable than FVIII alone; this interaction is important for effective hemostasis [910]. During FVIII maturation, calnexin and calreticulin promote FVIII folding; these interactions are important for FVIII protein quality control in the endoplasmic reticulum and Golgi apparatus [10]. In the case of a F8 gene mutation, truncation of the FVIII protein leads to dysfunction and disruption of its interactions with binding partner(s). Perturbations in these interactions predispose affected individuals to continuous bleeding.
F8 genotype profiling methods are well established [111213], facilitating identification of the mutation type(s) associated with the disease as well as the molecular interactions of HA. However, little is known about the relationship between F8 genotypes and their expressed phenotypes. Furthermore, how specific F8 mutations influence FVIII activity and/or protein-protein interactions is unclear. To further understand these mechanisms, it is useful to generate polypeptides corresponding to FVIII domains that are detectable by domain-specific antibodies. Therefore, we cloned each domain of F8 from Hep3B hepatocytes and produced FVIII domain-specific recombinant proteins.
Go to : 
MATERIALS AND METHODS
Materials
The pET-28a(+) vector was purchased from Novagen (Madison, WI, USA), and the pGEX-4T-2 vector was obtained from Amersham Biosciences (Uppsala, Sweden). BamHI and XhoI were purchased from Fermentas (Vilnius, Lithuania), and T4 ligase was obtained from Roche (Indianapolis, IN, USA). Luria Bertani (LB) broth was purchased from Narae Biotech (Gunpo, Korea), and skim milk, yeast extract, and tryptone for the 2× Yeast Tryptone (2× YT) medium were obtained from Difco (Detroit, MI, USA). Agarose-immobilized glutathione was purchased from Pierce (Rockford, IL, USA), and Poly-Prep Chromatography Columns were purchased from Bio-Rad (Hercules, CA, USA). Reduced glutathione was purchased from USB Corp. (Cleveland, OH, USA). Anti-rabbit IgG, anti-FIX, and anti-His-tag antibodies were purchased from Santa Cruz (Santa Cruz, CA, USA). An anti-vWF antibody was purchased from Abcam (Cambridge, UK), and the anti-GST antibody was purchased from Invitrogen (Carlsbad, CA, USA). Other reagents were purchased from Sigma-Aldrich (St. Louis, MO, USA).
Construction of domain-specific FVIII polypeptide expression plasmids
Each FVIII domain was cloned from Hep3B hepatocyte complimentary deoxyribonucleic acid (cDNA). Purification of Hep3B cDNA was performed using an RNeasy Plus Mini Kit (Qiagen; Valencia, CA, USA). F8 domain-specific polymerase chain reaction (PCR) primers are shown in Table 1. PCR thermocycler conditions are as follows: 30 cycles of 94℃ for 15 seconds (sec), 60℃ for 15 sec, and 72℃ for 1 minute (min) using Taq polymerase (NEB; Ipswich, MA, USA). Each PCR product was digested with BamHI and NotI for subcloning into pGEX-4T, an N-terminal GST (glutathione-S-transferase)-tagging vector. For subcloning into the 6× His-tagging vector, pET28, BamHI and XhoI were used for the A1 domain, whereas BamHI and HindIII were used for the A2, A3, and C domains. Lastly, DNA sequencing was used to verify that each FVIII domain was successfully subcloned. The recombinant plasmids were introduced into E. coli strain BL21 (DE3), and transformed E. coli was grown in LB medium supplemented with ampicillin (100 µg/mL).
Expression of GST- or His-tagged FVIII domain polypeptides
Bacteria harboring fusion plasmids were seeded in 10 mL 2× YT medium containing 100 µg/mL ampicillin (for the GST-tagging vector) or kanamycin (for the 6× His-tagging vector). Cells were cultured at 37℃ until an optical density (at 600 nm; OD600) of 0.4 was achieved. To induce FVIII protein expression, isopropyl β-D-1-thiogalactopyranoside (IPTG) was added to the culture medium at a final concentration of 0.5 mM. The cells were incubated for 4 hours (hr) at 30℃. Then, cells were harvested by centrifugation at 3,000× g for 20 min at 4℃. Cells were then resuspended in lysis buffer (137 mM NaCl, 8.0 mM Na2HPO4·7H2O, 1.4 mM KH2PO4, 2.7 mM KCl, 1 mg/mL lysozyme, 5 mM DTT, 0.03% SDS, and 1% Triton X-100, pH 7.4), and the mixture was sonicated for six cycles of 10 sec each at 5 sec intervals. The lysates were centrifuged at 16,000× g for 5 min at 4℃. Lastly, the extract was applied to sodium dodecyl sulphate-polyacrylamide gel electrophoresis (SDS-PAGE) for immunoblot analysis. Expression conditions were optimized in terms of the IPTG concentration, induction temperature, and incubation duration to obtain a high recombinant protein yield.
Cell extract preparation and purification of recombinant polypeptides
For GST-tagged protein purification, agarose-immobilized glutathione was added to the lysates and incubated, with shaking, for 1 hr at room temperature. To remove unbound proteins, mixtures were centrifuged at 1,250× g for 1 min at 4℃, and the supernatant was discarded. The pellets were resuspended in a wash buffer (137 mM NaCl, 8.0 mM Na2HPO4·7H2O, 1.4 mM KH2PO4, and 2.7 mM KCl, pH 7.4) and then loaded onto the Poly-Prep chromatography column. The column was washed twice with 5 mL of wash buffer, and the retained GST-FVIII protein was eluted with a buffer consisting of 137 mM NaCl, 8.0 mM Na2HPO4·7H2O, 1.4 mM KH2PO4 (pH 8.0), 2.7 mM KCl, and 100 mM reduced glutathione (GSH). For purification of His-tagged polypeptides, cells were lysed in lysis buffer (50 mM sodium phosphate containing 300 mM NaCl, pH 7.0) using ultrasonication followed by denaturation in lysis buffer containing 8 M urea. Cells were then centrifuged at 3,000× g for 20 min at 4℃ to remove cell debris. Protein purification was conducted under denaturing conditions using immobilized metal chelate affinity chromatography on cobalt resin.
GST pull-down assay
To assess the binding activity of the recombinant polypeptides, GST-tagged polypeptides corresponding to FVIII domains were incubated overnight with either Benefix (recombinant FIX from Pfizer) or albumin-free standard human plasma at 4℃. Next, 100 µL 50% glutathione-agarose bead slurry was added to the mixture and incubated on a rotor for 6 hr at 4℃. The beads were washed three times with 1 mL ice-cold GST lysis buffer. After centrifugation at maximum speed for 1 min, the supernatant was discarded. The beads or the eluted proteins were analyzed using SDS-PAGE and immunoblotting.
SDS-PAGE and western blotting
SDS-PAGE was performed using 10% acrylamide gels. Gels containing separated proteins were stained with Coomassie Brilliant Blue solution. For western blot analysis, proteins were transferred to a polyvinylidene fluoride (PVDF) membrane using the Trans-blot semidry system (Bio-Rad, Hercules, CA, USA). Membranes were blocked with 4% skim milk solution and incubated with appropriate primary antibodies. Immunoreactive proteins were detected using a secondary antibody conjugated with horseradish peroxidase, and they were visualized using the enhanced chemiluminescence method.
Go to :
RESULTS
F8 cDNA preparation and expression of FVIII domain polypeptides
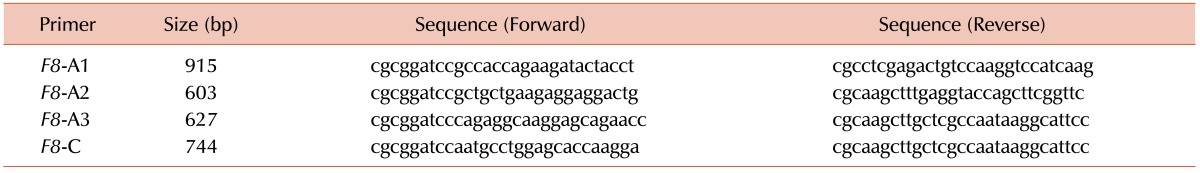
Human FVIII is normally expressed as a large glycoprotein containing the A1-A2-B-A3-C1-C2 domains. After activation by thrombin, FVIIIa transiently circulates as a heterotrimer consisting of A1, A2, and A3-C1-C2, and the B domain is released [14]. To prepare domain-specific DNA constructs from human F8, cDNA was synthesized from total RNA purified from the Hep3B cell line. Four primer pairs (Table 1) were designed to amplify F8 domain constructs corresponding to the A1, A2, A3, and C domains (Fig. 1A). Because of the relatively small size of the C1 and C2 domains, they were cloned together as domain C. Domain B was not cloned in this study because activated FVIII does not contain this domain [14]. Each DNA construct cloned into the pET-28a (+) vector was isolated and then subcloned into a protein expression vector. To confirm the fragment size, each construct was isolated from the pGEX4T-2 vector using BamHI and XhoI. The length of the F8 domain-specific DNA fragments was then determined. The following PCR products were obtained: 915 bp, 603 bp, 627 bp, and 744 bp, corresponding to the predicted lengths of the A1, A2, A3, and C domains, respectively (Fig. 1A and white arrowheads in Fig. 1B).
 | Fig. 1Cloning of each F8 domain. (A) A1, A2, B, A3, C1, and C2 represent the domains of human FVIII, and the bars indicate the cloning region of each domain. The numbers under each bar indicate the corresponding sequence of amino acids in each domain. (B) pET-28a(+) vectors containing each recombinant F8 domain were digested with BamHI and XhoI. Each domain was subcloned into pGEX4T-2, a GST expression vector. The plasmids were purified, and the products were subjected to BamHI and XhoI digestion. Lane 1, pGEX only; lane 2, pGEX-A1; lane 3, pGEX-A2; lane 4, pGEX-A3; and lane 5, pGEX-C. Arrowheads indicate the products of each domain. M1: 1 kb DNA marker, M2: 100 bp DNA marker.
|
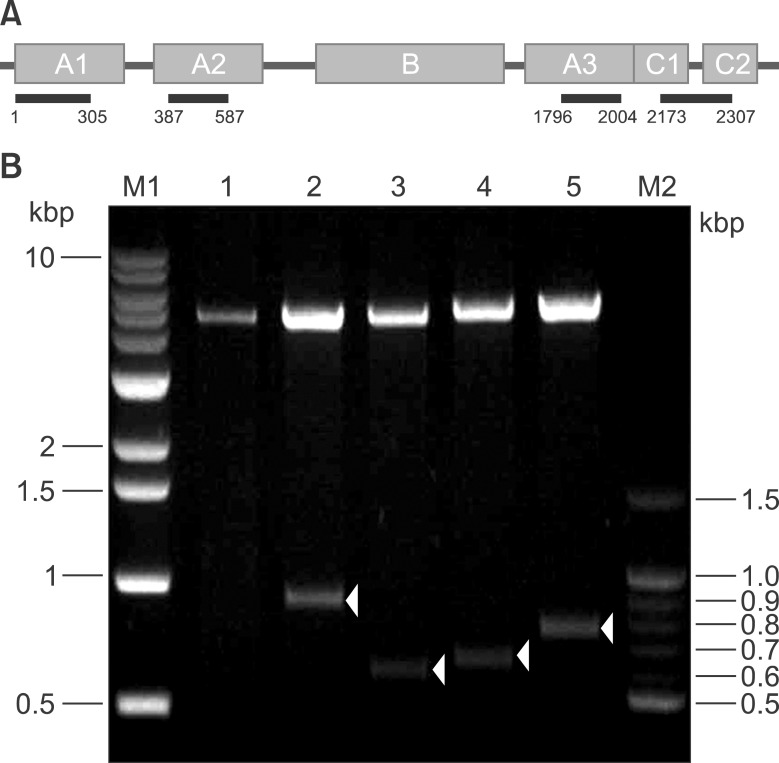
For expression of domain-specific recombinant polypeptides, the pGEX4T-2 or pET-28a (+) vectors were introduced into the E. coli BL21 (DE3) strain. Recombinant polypeptide expression was induced by adding 0.5 mM IPTG to the culture media for 4 hr at 30℃ in a shaking incubator. For analysis of GST fusion proteins, cell lysates were separated using SDS-PAGE and stained with Coomassie Brilliant Blue (Fig. 2A). Untransformed BL21 cells served as a negative control (BL21 in Fig. 2A). pGEX4T-transformed BL21 cells were used as a positive control for GST expression (Mock in Fig. 2A). In the mock control lane, corresponding to the empty pGEX4T-2 vector, 26-kDa GST protein bands were induced with 0.5 mM of IPTG (black arrowheads in the mock lane). Each F8 domain-specific fragment was expressed as a GST-fusion protein. The predicted sizes of GST-FVIII-A1, GST-FVIII-A2, GST-FVIII-A3, and GST-FVIIIC domains were 60, 50, 45, and 48 kDa, respectively (black arrowheads in Fig. 2A). To confirm expression of GST-tagged polypeptides, proteins from whole-cell lysates were separated using SDS-PAGE and subjected to western blotting using an anti-GST antibody (Fig. 2B). The anti-GST antibody did not bind BL21 cell lysates. In the mock control, a faint GST band was detected without IPTG, whereas expression of the GST protein was significantly increased upon addition of 0.5 mM IPTG (Fig. 2B).
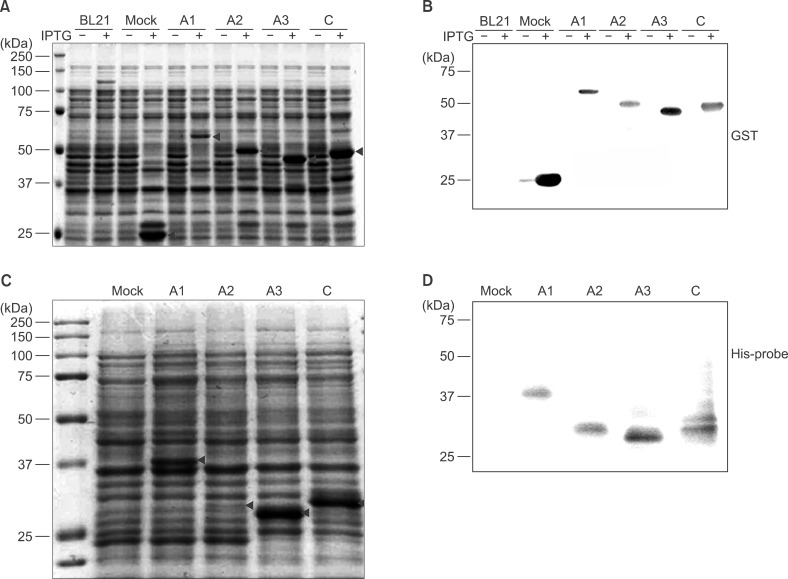 | Fig. 2Expression of recombinant FVIII polypeptides tagged with GST (A, B) or His (C, D). (A) Expression of recombinant FVIII polypeptides was induced by incubation with 0.5 mM IPTG for 4 hr at 25℃. To assess expression, whole-cell lysates were separated in 10% polyacrylamide gels. Gels were stained with Coomassie Brilliant Blue. (B) Proteins in the gel were transferred to a PVDF membrane and immunoblotted with an anti-GST antibody. BL21: host bacterial strain, Mock: pGEX4T-2 vector introduced into BL21. (C) To express His-tagged polypeptides corresponding to FVIII domains, the DNA fragment of each domain was subcloned into pET28a, a His-tagging expression vector. Expression of the His-tagged, domain-specific polypeptides was induced with 1 mM IPTG. (D) His-tagged polypeptides corresponding to FVIII domains were identified by western blotting using an anti-His-tag antibody. Arrowheads in (A) and (C) indicate the corresponding domain polypeptides.
|
His-tagged, domain-specific polypeptides were expressed from the pET-28a(+) vector. Expression was induced using 1 mM IPTG. The mock control consisting of the empty pET-28a(+) vector was the negative control (Mock in Fig. 2C). Each polypeptide of the His-tagged FVIII domain was separated using SDS-PAGE and subjected to immunoblotting using an anti-His-tag antibody. The polypeptides were identified by their predicted domain size (black arrowheads in Fig. 2C). The calculated size of A1, A2, A3, and C is 34, 23, 23, and 27 kDa, respectively. The polypeptides in Fig. 2C bound the anti-His-tag antibody (Fig. 2D).
Purification of recombinant polypeptides and binding characteristic validation
For recombinant polypeptide purification, bacteria expressing GST-tagged or His-tagged proteins were cultured until the OD600 reached 0.5, and then they were incubated with 1 mM IPTG. After optimization of GST-tagged domain-specific FVIII polypeptide expression conditions, protein fragments were purified using GST affinity chromatography.
For GST-tagged recombinant polypeptide validation, a GST pull-down assay was performed as described above. During blood clotting, coagulation factor IXa (FIXa) is known to bind FVIII via the A2 domain [1516]. Therefore, we tested whether each domain bound to recombinant FIX or platelet-derived FIX in standard human plasma. Results of the GST pull-down assay were shown by western blot analysis using the anti-FIX antibody. GST-tagged FVIII domain polypeptides were identified using the anti-GST antibody. All GST-tagged FVIII domain polypeptides were detected at the expected sizes among the pull-down products along with recombinant FIX (Fig. 3A and 3B). In contrast, recombinant FIX was only detected in complex with the FVIII-A2 domain polypeptide (indicated as rFIX in Fig 3A). The pull-down assay using standard human plasma also showed FIX in complex with the FVIII-A2 domain polypeptide alone (indicated as FIX in Fig. 3B). These results clearly showed that the synthetic polypeptides corresponding to FVIII domains have binding characteristics identical to neutral FVIII in human blood.
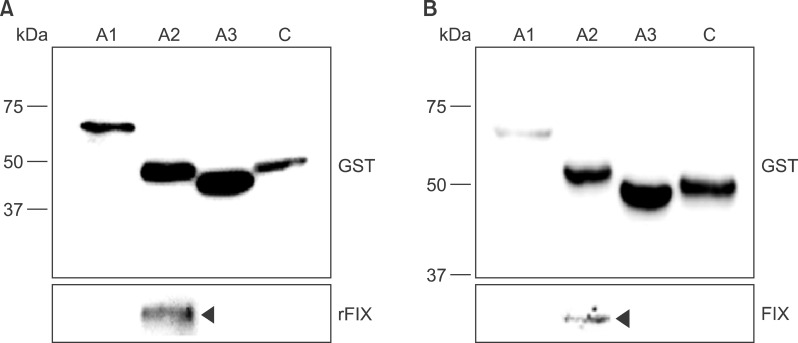 | Fig. 3Interaction of recombinant FVIII domain polypeptides with FIX. To characterize recombinant FVIII domain polypeptides, a pull-down assay was performed with FIX. GST-tagged polypeptides were purified and incubated with either recombinant FIX (A) or standard human plasma (B). The interacting proteins were precipitated by incubation with a 50% slurry of glutathione-agarose beads. The proteins bound to the GST-FVIII complexes were eluted and analyzed by western blotting with anti-GST (upper box) or anti-FIX (lower box) antibodies. Specific lanes: GST pull-down with GST-FVIII-A1 (lane 1), GST-FVIII-A2 (lane 2), GST-FVIII-A3 (lane 3), and GST-FVIII-C (lane 4).
|
Go to :
DISCUSSION
Owing to the recent work of several research groups, remarkable progress was made in understanding the molecular basis of HA. The method for profiling F8 genotypes is well established [111213]. This method is useful for identification of mutation type(s) associated with HA and its molecular interactions. However, the mechanism(s) that determine how F8 mutations influence FVIII activity and its protein-protein interactions remain unclear. FVIII is known to interact with diverse proteins, a property that may be important for effective hemostasis. Nonetheless, little is known about the types and functions of domain-specific FVIII binding partners. In this study, we prepared recombinant polypeptides corresponding to the A1, A2, A3, and C, domains, which can be useful tools for studies of FVIII domain-specific functions during blood coagulation.
In previous studies, cDNA generated from human leukocyte RNA was used to clone F8; however, the size of the PCR product contradicted with the predicted size [1718]. Thus, we conducted total RNA extraction from the human hepatoma cell line Hep3B. Our cDNA synthesis and PCR results were consistent with the predicted size of the F8 fragments. DNA sequencing was performed (Macrogen DNA sequencing service; Macrogen, Korea) to confirm the specific cloning of F8 DNA fragments. The data were analyzed using the Genomics Workbench program (CLC Bio; Aarhus, Denmark). Sequencing data from the cloned F8 fragments exactly matched the reference F8 sequence (GenBank accession # NM_000132.3; data not shown).
The FVIII domains were produced in E. coli, which is one of the most popular hosts for artificial gene expression. However, eukaryotic proteins can be defective when expressed in this system. In prokaryotic cells, the lack of eukaryotic chaperones and specialized post-translational modifications, such as glycosylation, can cause protein mis-folding and aggregation, resulting in insoluble protein-like inclusion bodies. To solve this problem, we subcloned each FVIII domain construct into the pGEX-4T vector (an N-terminal GST-tagging vector) to enhance the solubility of fusion proteins by using a solubility enhancing tag [19]. Natural FVIII is a glycoprotein. As expected, the recombinant A2 domain showed interaction with FIX (Fig. 3); however, the possible effects of the carbohydrate structure on protein-protein interactions should be further studied.
The main function of FVIIIa is to act as a protein cofactor during blood coagulation. The FVIII protein interacts with diverse proteins in human plasma. Because previous studies focused primarily on the coagulation process, studies regarding the specific roles of each FVIII domain are insufficient. In this study, we report an important step toward understanding the protein-protein interactions of FVIII during blood coagulation.
Go to :
 XML Download
XML Download