

 PDF
PDF ePub
ePub Citation
Citation Print
Print


INTRODUCTION
Embryonic stem (ES) cell or ES equivalent induced pluripotent stem cell (iPSC) are different from somatic stem cell such as hematopoietic stem cell (HSC) or bone marrow (BM) stem cell. ES or iPSC can be differentiated into all three germ layers such as endoderm, ectoderm, mesoderm and even germ cell that regenerate whole organism. Somatic HSC cells have limited differentiation ability lacking germ cell competency. Human embryonic stem cell technique brought the worldwide attention for therapeutic possibility of stem cell [1]. Those first hES lines from Thompson lab differentiated into three germ layers confirmed by SCID mice based teratoma assay. They are potent cells, however, the application of hES to real needed patients must overcome many barriers such as immune rejection, right tissue regeneration, cancer formation, and even ethical concerns to destroy human embryo. Somatic nuclear transfer (SCNT) mediated hES generation could be an alternative method to overcome patient specificity issue, however inefficient SCNT brought more ethical and technical concerns for use [2, 3]. A few years ago, Yamanaka lab found the way to avoid above mentioned hES or SCNT problem while introducing promising stem cell based transplant [4]. iPSC technique allows us to overcome immune rejection problem from allogeneic pluripotent stem cell based cell therapy. Recent STAP cell technology has brought worldwide attention and even lowered the huddle to enter regenerative medicine and iPSC field, confirming the velocity of regenerative research [5].
Nobel Prize winning iPSC technology has endowed us easy and ethical approaches to grasp human focused disease studies and regenerative medicine. Oct3/4, Sox2, Klf4, and cMyc mediated 4 factor Yamanaka's invention has improved since reported at 2007 [6]. However, original Yamanaka's 4 factors method [2, 4] is still a prominent option. iPSC is now exploding every sector of disease and academic part of research, we can use patient sample derived iPSC to test new therapy and study the undefined disease pathology. Herein, I would like to focus on recent progress and future outcome of iPSC derived personalized medicine and novel therapeutic options.
Go to : 
iPSC AS PLURIPOTENT STEM CELL
Since induced pluripotent stem cell (iPSC) technology has been invented to overcome the concerns and barriers to use human embryonic stem cells (hES), understanding hES would be proper before exploring iPSC [4, 6]. hES is immortalized population of inner cell mass from fertilized embryo - approximately 1 week after fertilization [1, 6]. Due to ethical barrier to generate normal embryo mediated hES, the genetically defected non-usable embryo has been used to generate hES for academic purpose [7]. Furthermore, aborted fetus derived human embryonic germ cell (hEG) were pursued as alternatives [8].
Various sources of mononuclear cells have been used to generate iPSC. Skin biopsy, hair follicle progenitor, muscle, bone marrows/mesenchymal stem cells, lymphocytes and even a few viable epithelial cells from urinal track have been used to generate iPSCs [4, 9]. Genetic and epigenetic memories from respected source of cells were issued in quality of iPSC, disease modeling, and cell based transplantation [10, 11, 12]. Bi-sulfide sequencing techniques revealed the vast differences in epigenetic markers between hES and iPSCs [13]. However, we don't know yet much about the meaning of chromosomal epigenetics difference and what those difference matters in biology. iPSC can be differentiated preferentially when differentiated into same tissue origin where iPSC originally came from [10, 14].
Although hES and iPSC have been quite well introduced and reproduced in various labs, there was a debate on whether hES or human iPSC (hiPSC) should be called embryonic stem cell like. There are reports indicating that hiPSC and hES are different in quality as mice embryonic stem cells are [15]. The hES and hiPSC have epiblast like shape and grow as colonies compared to mice ES cells, which form flat monolayer [16]. Thus, some groups started calling hES and hiPSC as epiblast stem cells [15]. Currently, there is no way to test the germline competent test for hES or hiPSC, which is the golden standard to test stemness. There is a recent publication studying non-rodent iPSC from pig generated by human iPSC factors [17]. The shape of porcine iPSC is similar to human ES or human iPSC, epiblast shape stem cell. The porcine iPSCs are germline segregated and can make transgenic pigs. Indirectly, porcine iPSC studies suggest that epiblast looking stem cell like morphology does not hinder them from becoming germline competent to make full organism [17, 18, 19].
Pluripotent stem cell based regenerative medicine could be the only cure for the patients with various degenerative diseases, spinal injuries, renal diseases, cardiac diseases, blood disorders and cancers [6]. Although allogeneic hES/iPSC derived differentiated tissues/cells has reduced level of MHC class II molecules, the allogeneic tissue rejection from hES mediated cell based therapy is one of the major challenges [20, 21]. Various trials and studies have been performed to overcome immune rejection from allogeneic hES derived tissues such as neurons or blood cell transplantation [21, 22]. Again, iPSC is competent enough to perform human disease modeling and tissue regeneration for cell transplantation [15, 23].
Go to :
DISEASE MODELING USING iPSC
hES has been studied and still used in many part of regenerative medicine as well as disease modeling. Before iPSC technique became popular, genetically defected embryos were studied for human disease such as Huntington disease embryo derived hES and fragile X-chromosome embryo derived hES [24, 25]. Disease hES mediated pathology modeling has unique advantages compared to animal studies and immortalized cell studies. The problem of difference in species often nullifies the novel drugs and therapy study results [26]. Immortalized cell based studies ignore pathology development issue in real patient. Patient derived pluripotent stem cell provides us unlimited access to fresh disease samples, and reliable novel therapy test and pathology studies with reduced side effects and off target effects.
There are many human diseases, which have not been recapitulated in small animal models, especially in adult onset diseases [26]. However, as we found handful amount of reports from hES derived human disease modeling, we looked for alternative method to continue human pluripotent stem cell mediated disease studies [24, 25].
Go to :
EARLY SUCCESS IN NEUROLOGICAL DISEASE MODELING USING iPSC
After Dr. Yamanaka succeeded his research, researchers envisioned disease modeling using hiPSCs and stocking up patient iPSCs library from 2008 [27]. The researchers studying the neurological disease modeling could generate patient astrocytes, neurons (motor neuron, peripheral neuron, etc.), and oligodendrocytes [28]. Subsequent reports were published, aiming most challenging human neurological diseases such as Alzheimer's disease (AD), amyotrophic lateral sclerosis (ALS), and Parkinson's disease (PD), which were hardly modeled by conventional rodent models [29, 30, 31]. The modeling studies were expanded to sporadic degenerative diseases, mostly diseases related to environmental stress origin (Table 1).
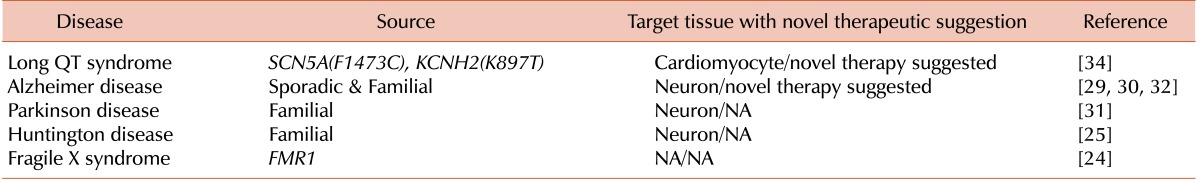
AD iPSC disease modeling has been reported by 3 different groups with various genetic background patients such as mutations in familial early onset AD related to presenilin 1 (PS1), presenilin 2 (PS2), and various forms of mutations in amyloid precursor protein (APP), even with sporadic AD cases [28, 29, 30]. Most recent AD iPSC modeling reported by Kondo et al. presented the subgrouping of two groups to show whether iPSC neurons had accumulation of Aβ oligomers inside of neurons or not [32]. One thing the study by Kondo et al. provides is that in iPSC adding DHA in culture as long as Aβ oligomer accumulation happens inside of neurons ameliorates both of familial and sporadic AD patient's symptom. That may explain why some AD patients respond to DHA treatment and the others do not, depending on AD pathology development. It suggests that the personalized medicine can be accomplished in near future, even for genetically non-identified human patient cases.
Go to :
PERSONALIZED MEDICINE CASE IN HEART DISEASE MODELING
The early onset childhood disease studies with stem cell based modeling have been very successful since stem cell based modeling recapitulates disease phenotype from patient iPSCs in early stage of differentiation [33]. In stem cell based culture, they showed reproducibility and distinguished disease phenotype within short time. One of the successful iPSC disease modeling cases is treating the heart disease.
Terrenoire et al. found the method of cure for early onset long QT (LQT) syndrome patients by simple change of heart beat rate [34]. It was the first reported iPSC modeling based living human disease treatment. LQT syndrome has 12 subtypes based on 12 different gene mutations. However, individual patient diagnosed as certain type of LQT syndrome has significant variations in clinical aspects as well as treating methods for these variations. Normally, Mexiletine was used as the first regimen and Flecanide was used for worse cases [35]. However, when the secondary Na+ channel blocker Flecanide was used, it blocked inside Ca2+ handling components, accompanying severe side effect [36]. LQT3 type (sodium channel mutation, SCN5A (F1473C)) patients did not respond to any known clinical regimen. As a result, there was no practical option for suffering patient when the study begun. Patient iPSC cardiomyocyte did not show any beneficial effect by adding Flecanide, confirming the patient's resistance to the conventionally used drugs [34].
Terreonoire et al. found that the simple heart beat increase in patient iPSC derived cardiomyocytes (CM) ameliorated the exacerbated Na+ activation without the use of Na+ channel blocker Flecanide [34]. Authors did not hesitate to apply the finding to suffering patient. When patient's heart beat rate was increased from 80 to 100 bpm, the patient obtained the most effective response to the treatment. This was the first case of personalized medicine using iPSCs disease modeling, which could not be accomplished by other disease modeling system such as heterologous SCN5A (F1473C) overexpression 293 cell lines or transgenic mice modeling studies.
Go to :
BLOOD DISORDER MODELING USING iPSC
Researchers require detailed knowledge of hematopoiesis in mutated blood stem cell to understand hematologic malignacies. Mutation in RAS pathway is commonly observed in leukemia and it has a complex pathway network which makes it hard to find right cure [37]. RAS activation in leukemia requires defined systemic approaches to test cancer-fighting treatment. Human disease studies have been performed with immortalized cancer cell lines or small rodent model, which could not cover the various mutations reported in real patients [26]. Human iPSC based disease studies could benefit us with more reliable treatment with more human focused manner. Patient derived iPSC blood lineage studies could be the game changer in hematopoietic disease modeling.
The initial blood disorder disease modeling has been focused in two major diseases, sickle cell anemia and leukemia [38, 39, 40, 41, 42]. More reports regarding blood disorder iPSC disease modeling are summarized in Table 2. It is not surprising to find blood disorder disease modeling samples originated from defected blood iPSCs. Initial trials for iPSCs disease modeling of hematopoietic disorder, scientists used blood stem cell such as bone marrow (BM), cord blood (CB), BM derived MSCs for generating iPSC [41]. Highly proliferating somatic blood stem cells rather than fully mature cells are good source for reprograming. However, BM needs mobilization and CB needs to be cryopreserved for long periods before disease occurs. Current technical advance in mature blood cell based iPSC generation allows more researchers use patient blood samples instead using fibroblast, BM or CB [43, 44].
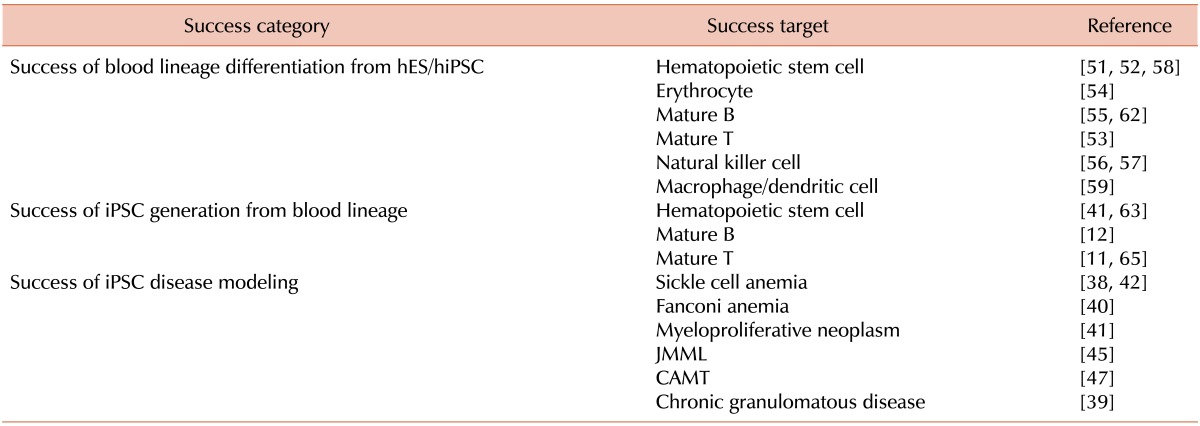
Ye and colleagues reported the JAK2α-V617F mutation containing iPSCs from Myeloproliferative neoplasm (MPN) patient [41]. They used both CB and BM (CD34+) cells to generate iPSCc and subsequently differentiated into blood progenitors (CD34+CD45+). Myeoloproliferative neoplasmic JAK2α mutated iPSC showed dramatic increase of proliferative activity and increased portion of erythroid, which recapitulate affected MPN patient pathology [41]. Raya et al. also provide iPSC disease modeling from Fanconi anemia blood disorder patient [40]. Novel therapeutic options nor drug test were not performed at early leukemia iPSC modelings [40, 41], however, several attempts to test gene editing with sickle cell anemia iPSC modeling have been reported [38, 42].
More aggressive proof of concept iPSC disease modeling evolved recently. Juvenile myelomonocytic leukemia (JMML) iPSC disease modeling showed more advanced feature of blood disorder iPSC disease modeling [45]. Using independent two male JMML patient blood samples containing PTPN11 (E76K) missense mutations, researchers generate iPSCs. Hematopoietic lineages differentiated from iPSC were used and tested for proof of concept drug screen. From JMML patient derived iPSC, researchers found that activated MEK and JAK1/2 pathway could be a target for future treatment [45].
Hirata and colleagues constructed congenital megakaryocytic thrombocytopenia (CAMT) patient derived iPSC to perform CAMT disease modeling, which hardly recapitulated with conventional small rodent model [46]. CAMT disease has mutations in MPL gene (thrombopoietin receptor) and significantly reduced platelets and megakaryocytes. Mice with MPL homozygotes knockout does not recapitulate conventional CAMT human disease with absence of megakaryocytes indicating that the pathophysiological development might differ from human and mice [47]. The only treatment for CAMT patient is allogeneic bone marrow transplantation. The advantage to use iPSC disease modeling is that the researcher can monitor the disease development and pinpoint the critical pathological development, which can be used for finding new therapy. Hirata and colleagues found patient iPSC derived hematopoietic differentiation ended up less than 1% of platelets number compared to that of normal iPSC. Retroviral introduction of normal MPL gene in defected iPSC caused another findings regarding platelets biology. Fli1 gene activity is greatly increased in CAMT iPSCs derived blood progenitors compared to normal iPSC groups, indicating possible down stream pathway to bring novel approaches to cure CAMT.
Go to :
BENEFIT TO USE iPSC IN BLOOD CELL REGENERATION AND CELL BASED THERAPY
In conjunction with hES based studies, we can categorize the usage of iPSC technology into two major parts: First, human disease modeling which has advantage in pathogenic studies especially in neurodegenerative and cardiac disorder field; Second, patient specific cell-based therapy with iPSC derived regenerated tissue. Now researcher can use human focused study to find human treatment therapy. Patient specific tissue generation from patient's own cell mediated iPSCs makes perfect match for transplantation without severe immune suppression regimen or possible allogeneic tissue immune rejection, although hES/iPSC derived tissues are less immunogenic [21].
Stem cell based cell transplantation is not a new concept in hematological research community. Using hematopoietic stem cell transplantation (HSCT) to reconstitute dysfunctional blood system has been widely performed for last four decades. HSCT is a potent therapeutic regimen for leukemia, sickle cell anemia and even in cardiac disease patient. HSCs from adult bone marrow and umbilical cord blood cells have been major curative modality for treating blood disorders including leukemia, sickle cell anemia, granulomatous anemia [39, 48, 49]. Historically, cell based therapy has been routinely mentioned in the regenerative medicine field and largely fortified by successful HSCT [49]. However, there are known obstacles in blood stem cell based therapy such as challenge to find the right allogeneic donors and graft versus host diseases. Also limited quantity of allogeneic HSC may hinder the success of HSCT. Autologous HSC is another method that is used, however, it also has its own limitations. A few trials with autologous transplant using retro/lenti-virus insertion based genetic alternation on patient's hematopoietic stem cell have been applied as an alternative [50]. A case of autologous HSCT with retroviral mediated γc cytokine receptor in an 11 year old patient with X-linked severe immune deficiency raised a great hope in autologous gene therapies, however, it had faced danger of leukemia by unexpected viral genome insertion [50]. Also, lack of tools to expand autologous populations of HSC lowers HSC transplantation success rate. Instead of using rare somatic HSC donor cells, scientists began looking for alternatives to avoid rarity issue of HSC.
In 2001, Kaufman et al., succeeded in the differentiation of blood cells derived from non-hematopoietic stem cells using hES [51, 52]. Since then, various types of blood progenitors and blood lineage cells were differentiated from human pluripotent stem cell to B cells, T cells, NK cells, macrophages, erythrocytes in more defined and efficient way [49, 53, 54, 55, 56, 57, 58, 59]. Furthermore, mouse ex-vivo studies confirmed the efficacy and potency of mice iPSC derived blood cells and their functional recovery in irradiated mice studies, confirming the efficacy and safety issue in future human trials [60, 61].
By changing the source of stem cell from hES to patient specific iPSC, we can obtain compatible quality of blood lineage cell differentiation for future human therapy [56, 62]. Furthermore, by using iPSCs, we can overcome two major obstacles which hES mediated hematopoietic transplantation therapy has. Using the patient's autologous stem cell generations, immune rejection problem could be abolished. The patient specific genetic/epigenetic memory would be useful traits for disease targeting blood cells.
Human blood cells are the most accessible human samples in general clinics. It is usual procedure to characterize patient blood with biochemical, histological and genetic methods in clinics [11, 63]. Patient may not prefer skin biopsy, which accompanied with unnecessary bleeding, discomforts, permanent scars and infection. Hair follicles and urine sample iPSC generation may come with bacterial contamination hindering future cell based therapy. Small portion of peripheral blood cells is enough to generate iPSCs with conventional Yamanaka factors [63]. Even small number of mature blood cells such as B, T, and NK T Cells have been successfully transformed into germ line competent iPSCs [11, 12, 63, 64, 65].
Go to :
BLOOD DERIVED iPSCs AND TISSUE SPECIFIC MEMORY
Most successful blood samples for generating iPSC are umbrical cord blood, which contains most of highly proliferating immature blood cells such as CD34+ or CD133+ cells [63]. Previous success story even in peripheral blood iPSC generation was believed to have arisen from CD34 and CD133 progenitor cells. Lineage specific approaches have been reported such as in B and T cells [11, 12, 63, 64, 65].
One thing unique in lineage specified blood cell derived iPSCs is that they contain original memory. Mature B cell has reassembled V (variable), D (diversity), and J (joining) segments and derived iPSC showed same fingerprint as original B cell [12, 64]. Mature T cell also shared same TCR rearrangement pattern after transformation into iPSC [11]. Besides apparent genetic alteration from the source of iPSC cells, epigenetic advantages to become iPSC origin tissue have been reported [10, 14].
In theory, if a person has certain disease such as cancer or infectious disease then the patient's immune cells contain most of the pathogen fighting immune cells. By converting and expanding those diseases fighting cells into iPSCs, we can harness therapeutic advantage. It is very tempting hypothesis that iPSCs derived from suffering patients might fight back when re-differentiated into disease fighting cells such as B, T, NK, and macrophages (Fig. 1).
 | Fig. 1Patent specific iPSC disease modeling and cell based transplantation therapy. Mononuclear cells from patient are used for generating iPSC. Depending on the source of cells, both memory contained cells such as mature blood cell and memory lacking immature cell mediated iPSC are generated. Subsequent differentiation from patient specific iPSC can be directly used for personalized cell based therapy with proper cell lineages such as blood, muscle, and neuron. Gene editing technology such as ZFN, TALEN, and CRISPR will be utilized to fix the genetic error from affected patients even with large DNA deletions. Also, patient iPSC based personalized medicine to find right dosage of known medications or novel therapeutic agents can be tested.
|
Go to :
GENE EDITING AND GENE THERAPY BASED iPSC DERIVED PERSONALIZED MEDICINE
Recent progress in gene editing and gene delivery technology allows us to fix or insert defected mutations without inserting or adding unnecessary viral gene or non-human genomes [66, 67](Fig. 1). Without the establishment of global iPSC banking, it is hard to find non-immunogenic iPSC donors from allogeneic source. However, if the patient can use autologous cells to make iPSC, then those iPSC derived blood cells and tissues can be used without severe immune suppression regimen. Zinc finger nuclease (ZFN) is the fused construct of zinc finger head and fok1 nuclease [67]. ZFN can specify 3 consecutive nucleotides with single zinc finger domain. Usually, ZFN comes up with 3 or more domain combinations to gain DNA binding specificity [67]. Transcription activator-like effector nuclease (TALEN) derived from plant bacteria DNA binding domains can specify single nucleotide with 30-35 amino acids repeats [68]. However, elongated tandem repeats hinder huge construct to manipulate specific target sequence. Contrast to ZFN and TALEN, clustered regularly interspaced short palindromic repeats (CRISPR) use only small size of RNA fragment to select and dissect target DNA sequence [69]. In all three cases of gene editing, they make double stranded DNA breaks, which require homologous DNA donor to replace as part of eukaryotic DNA repair system. Details of gene editing without tracing of viral or non-human genome technology have been well reviewed in recent reports [67, 69].
One interesting proof of concept approach is suggested by Zhou et al.'s X-linked chronic granulomatous disease based study [39]. They generated mutated gp90phox patient specific iPSC then inserted normal gp90phox gene in AAVS1 locus at human chromosome 19 [39]. AAVS1 locus has been considered for safe harbor for exogenous gene insertion [70]. Large deletion cannot be rescued by single nucleotide fixation like ZFN, TALEN, and CRISPR-Cas. By combining site-specific nuclease activity from TALE or Zinc finger, scientist can insert large size of gene into patient iPSC to rescue defects.
Go to :
CONCLUSIONS
The modern technique to regenerate personalized iPSC changes our way of thinking in therapy development. Now we can test novel therapeutic options with samples from patients without limit. We can even regenerate needed tissue such as patient matching blood, muscles and neurons. Although it is a quite recent technology, iPSC disease modeling can be acclaimed to be close to certain therapeutic success. Stem cell transplant in clinics is not new and we have decade's history of successful HSCT. Simply changing the source of transplant from allogeneic HSC to autologous iPSC, we are cautiously expecting another breakthrough in human medicine. Patient specific drug treatment and cell based transplant with needed tissues are both personalized medicine. These two main approaches to cure human disease using human iPSC are equally important and relevant. Which achievements would come to first? That will be fascinating question.
Go to :
 XML Download
XML Download