

 PDF
PDF ePub
ePub Citation
Citation Print
Print


TO THE EDITOR: Pure erythroid leukemia (PEL) has been defined as a rare, aggressive disease characterized by a neoplastic proliferation of immature cells committed solely to an erythroid lineage with no increase in myeloblasts in the bone marrow (BM) [1, 2, 3]. According to the 2008 World Health Organization (WHO) classification [4], PEL is classified as acute myeloid leukemia (AML), not otherwise specified, only when the case does not fit into any other specific categories. PEL is defined by the presence of immature erythroblasts, which should comprise at least 80% of the BM cells, with no evidence of a significant myeloblastic component [2, 3, 4]. PEL accounts for about 10-20% of all acute erythroid leukemias (AEL) and less than 1% of all AML cases [2], and it has been very rarely reported as a therapy-related AML [5, 6, 7, 8]. Moreover, its occurrence has never been reported after exposure to chemotherapy and radiation for breast cancer.
Here, we describe a case of a 64-year-old woman with a long history of heavily treated breast cancer. She was referred for hematological consultation after failure of erythropoietin treatment for anemia. For the past 16 years, the patient had received multiple courses of radiotherapy and several chemotherapeutic and endocrine agents for her breast cancer. The patient was previously diagnosed with left breast cancer (invasive ductal carcinoma) with involvement of the axillary lymph nodes (pT1b, pN1 [1/6], M0, stage IIA). Cancer cells were positive for estrogen receptor (ER) and progesterone receptor (PR); human epidermal growth factor receptor (HER2) was not tested at that time. The patient was treated with surgery (quadrantectomy and axillary lymph node dissection) and adjuvant chemotherapy (6 courses of CMF: cyclophosphamide 600 mg/m2, methotrexate 40 mg/m2, and 5-fluorouracil 600 mg/m2; cycle repeated every 3 weeks) followed by adjuvant tamoxifen (20 mg/day). The first recurrence was observed 4 years later at a distant site, the T12 vertebra. After receiving radiation therapy (39 Gy in 13 fractions) to the dorsolumbar spine (T11-L1), the patient achieved a complete remission; therefore, monthly zoledronic acid and daily aromatase inhibitor exemestane were given as maintenance therapies. However, a second relapse (nodal involvement in the right axilla) occurred 3 years later (7 years after initial onset) in the form of an invasive triple negative (negative for ER, PR, and HER2) ductal carcinoma. Thereafter, the patient received docetaxel (100 mg/m2 every 3 weeks for 6 cycles) along with local radiotherapy (40 Gy in 20 fractions), achieving a complete remission. However, 2 years later (i.e. 9 years after the first diagnosis), a third tumor relapse occurred. The patient presented with liver metastasis, multiple adenopathies (para-aortic), and diffuse bone osteolysis involving the left fourth rib, the T12 vertebra, and the right iliac wing. Thereafter, non-pegylated liposomal doxorubicin (60 mg/m2) combined with cyclophosphamide (600 mg/m2) was administered for 6 cycles (each cycle every 3 weeks), and a partial response was achieved. Next, to maintain long-term disease control, the patient received a cyclophosphamide-based metronomic regimen of cyclophosphamide and methotrexate (CM; cyclophosphamide 50 mg p.o. daily and methotrexate 2.5 mg p.o. twice daily on days 1 and 4). However, because of further local disease progression, the patient received radiotherapy targeting the newly recurrent hepatic lesions (75 Gy in 3 fractions) and abdominal lymph nodes (45 Gy in 6 fractions). The CM protocol was interrupted because of additional hepatic metastases and other multiple neoplastic localizations; in particular, a pleural mass and effusion in the left hemithorax, and multiple osteolytic lesions involving the left fourth rib, the left acetabulum, and the ipsilateral iliac wing were detected. Thereafter, the patient received additional radiotherapy (20 Gy in 5 fractions) targeting the skeleton and thorax mass. Next, palliative chemotherapy with vinorelbine (60 mg/m2, days 1, 8, and 15 every 3 weeks) combined with capecitabine (1,000 mg/m2, days 1-14 every 3 weeks) was administered for approximately 6 months.
When the patient came to our medical facility, she had advanced and active disease with pleural effusion, moderate liver injury, and multiple and extensive bone lesions. She had been receiving erythropoietin (10,000 IU thrice a week) for 3 months because of anemia considered to be a secondary cytotoxic effect of ongoing chemotherapy. The erythropoietin stimulation allowed only for a transient benefit, and the patient continued to receive occasional transfusions. At physical examination, she was pale and fatigued with some petechiae. Hepatosplenomegaly was also noticed. Laboratory data revealed marked anemia with low reticulocyte count and thrombocytopenia. Serum levels of lactate dehydrogenase and ferritin were markedly elevated, and direct and indirect Coombs' tests were negative. Vitamin B12 and folate blood levels were in the normal range. Renal function was normal, and hepatic parameters, such as alanine aminotransferase and aspartate aminotransferase levels, were only slightly altered. A coagulative analysis profile revealed no abnormalities. Serology for human immunodeficiency virus, hepatitis virus, cytomegalovirus, Epstein-Barr virus, and parvovirus B19 were negative. The morphological examination of peripheral blood smears showed marked anisopoikilocytosis and thrombocytopenia, abnormal granulation of neutrophils, but not circulating blasts. The BM smear was hypercellular with prominent erythroid proliferations (65% of total BM nucleated cells) and megakaryocytic dysplasia; a slight increase in proerythroblasts and basophilic erythroblasts was observed, with a left shift in the erythroid cell population, although normal developmental erythroid stages were still recognizable. The examination of the BM trephine biopsy confirmed these findings, with 65% of erythroid cells positive for Glycophorin A (Glyc-A) and CD34 but negative for myeloperoxidase (MPO). CD34-positive myeloblasts were less than 1%. Cytokeratin staining with MNF116/AE1AE3 was negative. Cytogenetic evaluation revealed complex karyotype abnormalities in all of the metaphases: 44,XX,der(12)(p?→qter), der(14)t(14;21)(q11.2;q11.2),der(16)t(16;21)(q11.2;q11.2), der(17)t(17;21)(p12;q11.2),der(19)t(19;21)(p13.3;q11.2),-21,-21. Fluorescence-activated cell sorting immunophenotype by flow cytometry (FC) was not performed because of the insufficient amount of BM aspirate. This scenario was interpreted as therapy-related myelodysplastic syndrome (MDS) (refractory anemia) with erythroid predominance, and given the high transfusion burden, the patient received erythropoietin at MDS doses (40,000 IU twice a week); however, given the lack of efficacy, erythropoietin was subsequently withdrawn. Thereafter, in the following 2 months, the transfusion requirement of red blood cells and platelets further increased. A new BM aspiration resulted in only scarce BM material in BM smears. Despite these limitations, the cytomorphology was consistent with a substantial progression of MDS-related features and a predominance of erythroid immature elements (Fig. 1). The BM biopsy (Fig. 2) revealed prominent MDS features, including a marked increase (more than 90% of nucleated cells) of atypical erythroid precursors and, in a lesser amount (10-15%), immature megakaryoblasts. Myeloblasts (CD34-positive cells) were < 5% of the total BM population. The erythroid precursors showed a markedly aberrant morphology; this finding, along with a concurrent immature megakaryocytic population, suggested an erythroid-megakaryocytic neoplastic BM infiltration. The immunophenotypic analysis by 3-color FC with CD45/side scatter gating demonstrated that immature proerythroblasts expressed HLA-DR, CD36, CD44, and Glyc-A, whereas other markers (anti-MPO, CD2, CD3, CD4, CD7, CD13, CD14, CD15, CD19, CD22, CD33, CD34, CD38, CD45, CD56, CD64, CD79,CD117) were negative. The hypodiploid complex abnormalities found at the initial MDS diagnosis were confirmed. Molecular studies for the most frequent AML-related alterations (NPM-1 and FLT3-ITD mutations; AML1/ETO, CBFβ/MYH11, PML/RARα, and MLL rearrangements) showed no abnormalities. Unfortunately, mutational analysis was not done at this time. Thereafter, the patient was diagnosed as having a therapy-related AML, according to WHO 2008 classification [4]. However, given the predominant (>80%) erythroid population and the near absence of myeloblasts in the framework, a secondary diagnosis of PEL was made. Given her poor clinical status, the patient was unsuitable for induction chemotherapy and died 2 months later of refractory gastrointestinal bleeding and pneumonia.
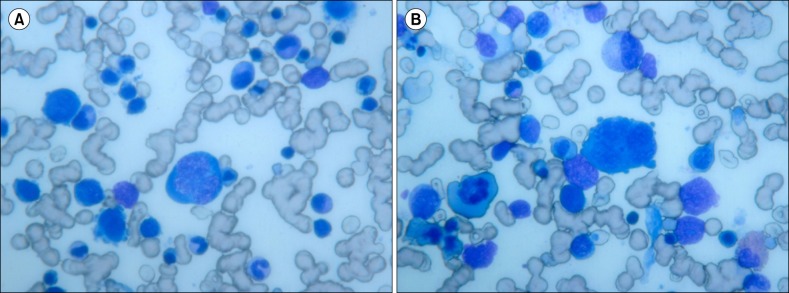 | Fig. 1Bone marrow smears (×400, panoptical staining) showing several erythroblasts in different maturation phases with clear dyserythropoietic features (maturative asynchronism, cytoplasmic blebs). (A) A bone marrow smear revealed megaloblastoid proerythroblasts with polychromatophilic cytoplasm, multiple cytoplasmic blebs, and polylobated hyperdiploid nucleus with finely dense chromatin. (B) A bone marrow smear revealed giant proerythroblast with scarce basophilic cytoplasm and round nucleus with immature chromatin.
|
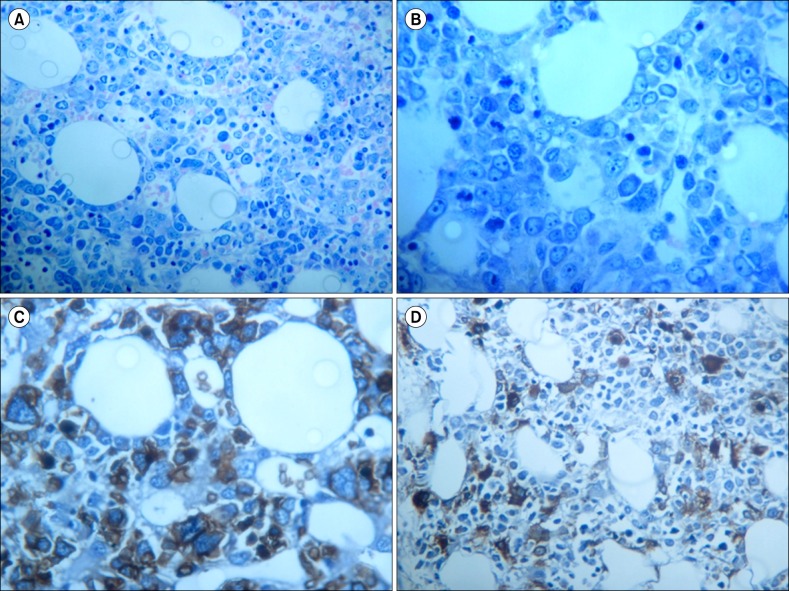 | Fig. 2Bone marrow trephine biopsy at diagnosis of pure erythroid leukemia. (A) Medium magnification (×200) shows a predominant bone marrow population composed of atypical immature erythroid precursors (more than 90% with nucleated cells). (B) High magnification (×400) revealed numerous atypical erythroid precursors and dyserythropoiesis. (C) Glycophorin-A immunostain expressed by the atypical erythroid neoplastic precursors. (D) Linker for activated T-cells immunostain shows dysmegakaryocytopoiesis with a micromegakaryocytes and a population of immature megakaryoblasts.
|
An increased risk of developing therapy-related AML, including AEL [9], in breast cancer patients due to exposure to radiotherapy and/or adjuvant chemotherapy regimens has been recognized [9]. However, PEL as a consequence of chemoradiotherapy in the setting of breast cancer has not been previously reported. Since leukemic erythroblasts have variable immunophenotypes and lack clear lineage-defining markers, this occurrence imposes great diagnostic challenges to pathologists and necessitates a careful and open-minded approach because left-shifted erythroid maturation can occur in various neoplastic and non-neoplastic conditions. In particular, in our case, erythropoietin stimulation would be a possible cause of the erythroid hyperplasia; however, this hypothesis was ruled out because at the time of worsening of the hematological situation that prompted the PEL diagnosis, our patient had not received erythropoietin for the last 8 weeks. Our diagnosis relied only on conventional findings and traditional tools; unfortunately, we did not pursue further testing on the molecular abnormalities related more specifically to erythroid and therapy-related dysplastic/leukemic abnormalities (DH1/2, TET2, ASXL, EZH2, or the expression profile of transcription factors such as GATA-1) that could have provided more information on an already complex genotype [10].
In conclusion, we present a new case of PEL, characterized by the massive proliferation of proerythroblasts and a markedly complex karyotype, with a very poor outcome in an advanced breast cancer patient.
 XML Download
XML Download