

 PDF
PDF ePub
ePub Citation
Citation Print
Print


INTRODUCTION
Castleman's disease (CD) is a rare, polyclonal, lymphoproliferative disorder first described by Dr. Benjamin Castleman in 1954 [1]. Originally, unicentric forms of CD were reported as a single, mediastinal mass with systemic symptoms that could be resolved after surgical excision [2]. On the other hand, patients with multicentric CD, defined by the involvement of at least 2 noncontiguous, lymph node areas, were often refractory to treatment and showed worse clinical outcomes [3, 4, 5]. Because multicentric CD is rare, there are no established treatment guidelines, standardized diagnosis criteria, or well-known prognostic factors. Furthermore, although clinical presentations, treatments, and outcomes of human immunodeficiency virus (HIV)-associated multicentric CD have been studied more extensively [6, 7, 8, 9], there are not enough data regarding patients without HIV infection. A few studies of patients with HIV-negative, multicentric CD attempted to analyze the clinical characteristics of CD and correlate variables with clinical outcomes; however, there is no well-established system for assessing the prognosis.
We performed a retrospective analysis of the clinical factors in Korean patients with HIV-negative, multicentric CD to analyze the prognostic implications of those factors.
Go to : 
PATIENTS AND METHODS
Patients with pathologically proven CD were retrospectively identified from the clinical database of the Asan Medical Center, Seoul, Korea. Eighty patients were diagnosed with CD between January 1990 and March 2013. Of these, 32 patients had multicentric CD. Four patients who did not undergo an HIV serology test were excluded; as a result, 28 HIV-negative patients with multicentric CD were included in this analysis. The extent of disease was defined as involvement of a single side and of both sides of the diaphragm as seen on computed tomography, positron emission tomography, and/or positron emission tomographycomputed tomography. At analysis, all pathologic diagnoses were reviewed by an expert hematopathologist (J. Huh) and reclassified. Human herpes virus-8 (HHV-8) was tested for with immunostaining against the HHV-8-latent nuclear antigen (Cell Marque Corp., USA) or by PCR for HHV-8 DNA detected in pathological sections.
Overall survival (OS) was calculated from the diagnosis to a patient's death from any cause. Data were censored if patients were still alive at the last follow-up. Categorical variables were evaluated using the chi-squared or Fisher's exact test, as appropriate. Survival curves were plotted according to the Kaplan-Meier method, and the log-rank test was used to evaluate the prognostic significance of the clinicopathologic characteristics. All statistical analyses were performed using the Statistical Package for the Social Sciences (SPSS, Chicago, IL, USA), version 21.0, with 5% defined as the level of significance.
Go to :
RESULTS
Baseline patient characteristics
Baseline patient characteristics are described in Table 1. The median age was 54 years (range, 13-75), and 22 patients were male. All patients had been diagnosed since 2000, except for 2 who were diagnosed in the 1990s. The hyaline vascular variant (HVV) (N=11, 39%) of CD was the most frequent histologic subtype. Frequently observed signs and symptoms seen at diagnosis included hepatomegaly or splenomegaly, fever, edema, ascites, and pleural effusion. Sixteen patients (57%) had disseminated disease (i.e., involvement of both sides of the diaphragm), whereas 11 (39%) patients had limited disease (involvement of a single side of the diaphragm). In 1 patient, disease extent could not be exactly estimated because imaging studies for the abdomen and pelvis were not performed.
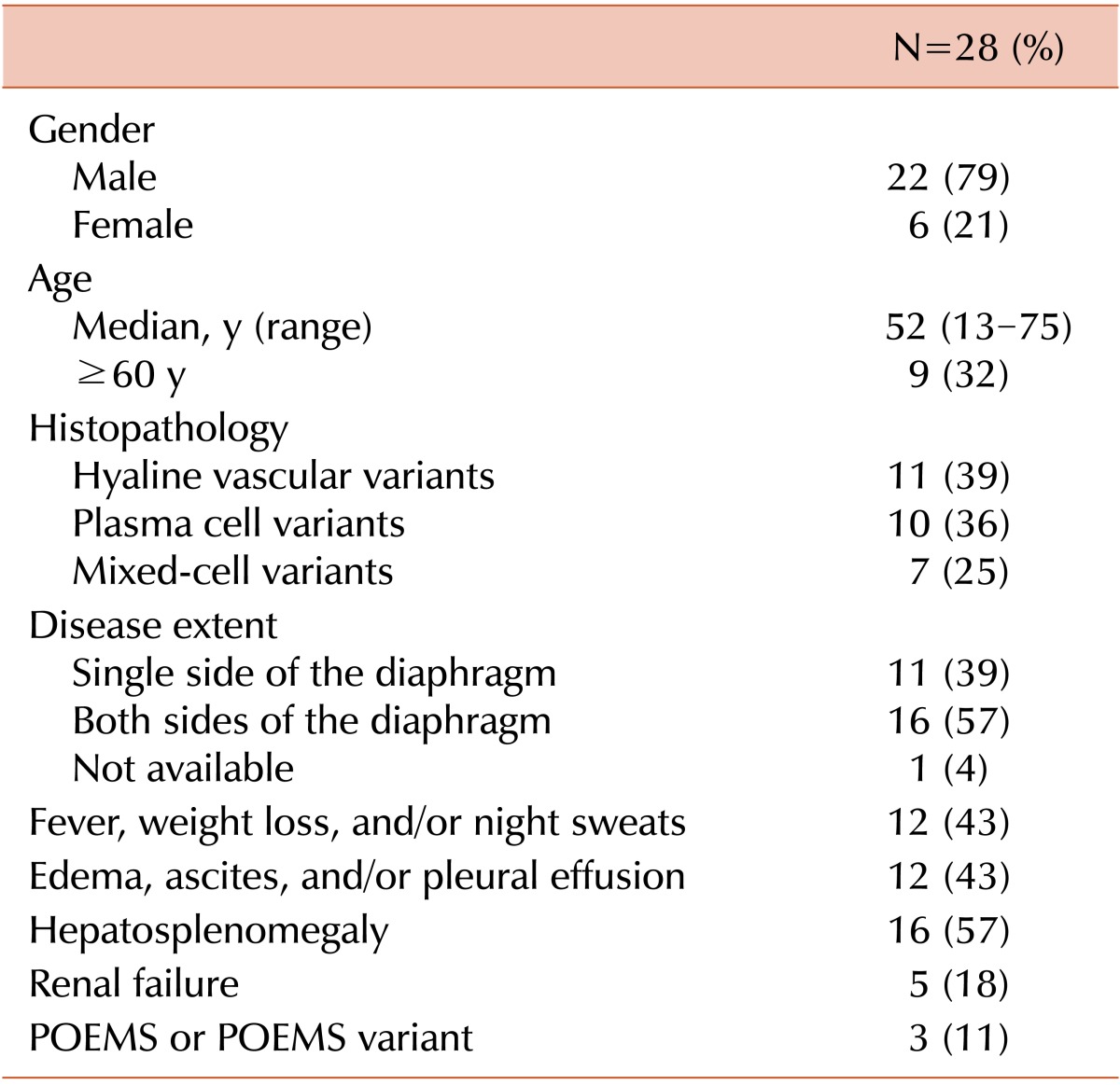
One patient had concurrent polyneuropathy, organomegaly, endocrinopathy, monoclonal plasma-proliferative disorder, and skin changes (POEMS) syndrome accompanying polyneuropathy, IgG λ monoclonal gammopathy, splenomegaly, edema, skin pigmentation, and adrenal insufficiency. Two other patients had monoclonal gammopathy, each with IgM λ and IgA λ, but not accompanying polyneuropathy. Despite the absence of 1 mandatory diagnostic criterion, these 2 patients had other features of POEMS syndrome, including extravascular volume overload, organomegaly, and endocrinopathy (adrenal insufficiency and/or pituitary insufficiency). According to a recently proposed concept, these patients might be classified as having a CD variant of POEMS [10]; therefore, 3 patients (11%) in our study cohort had related POEMS syndrome or its variants. One patient had a history of Kaposi's sarcoma treated with radiation therapy.
The laboratory results obtained at diagnosis are summarized in Table 2. Nineteen of 27 tested patients (70%) had anemia. An antiglobulin test was conducted in 8 patients with anemia, and 7 (87%) were positive for the assay. One showed elevation of the reticulocyte productive index and decreasing haptoglobin, both of which were consistent with autoimmune hemolytic anemia. This patient also had autoimmune thrombocytopenia, leading to the diagnosis of Evans syndrome. Thrombocytopenia was also observed in 30% of the other patients. C-reactive protein and erythrocyte sedimentation levels were elevated in 83% and 93% of patients, respectively, and lactate dehydrogenase and β2 microglobulin levels were elevated in 52% and 76% of patients, respectively.

Bone marrow examination was performed in 19 patients, 10 of whom (53%) showed reactive plasmacytosis. One patient fulfilled the criteria for asymptomatic multiple myeloma. Three patients tested positive in the PCR assay for HHV-8 among the 10 tested. Four patients were examined with tissue immunohistochemistry (IHC), and 1 patient was positive for HHV-8. Two of these patients underwent both of the tests; 1 showed a negative result on both tests, whereas the other was positive in the PCR assay but negative in the IHC test.
Extravascular volume overload was significantly associated with several clinical factors, including disseminated disease extents (P=0.01), and constitutional symptoms-fever, weight loss, and/or night sweats (P=0.006), organomegaly (P=0.02), and hypoalbuminemia (P=0.04).
Treatment and outcomes
Best responses to initial therapy are summarized in Table 3. As the first-line treatment, cytotoxic chemotherapy was administered to 11 patients (39%); the most common regimen was a cyclophosphamide, doxorubicin, vincristine, and prednisolone combination (CHOP, N=7). Among 7 patients who received CHOP, 5 completed 6 cycles of chemotherapy, 1 was transferred to another hospital after 2 cycles, and 1 was lost to follow up after 1 cycle. Other patients were treated with cyclophosphamide, vincristine, and prednisone (CVP; without doxorubicin from CHOP, N=3) and rituximab plus CHOP (N=1). Among those who underwent CVP chemotherapy, rituximab was added from the second to sixth cycles in a patient with Evan's syndrome (N=1). The other 2 patients who were treated with CVP received 3 cycles and 5 cycles, respectively. In a patient with POEMS syndrome, 1 cycle of rituximab plus CHOP was followed by 6 cycles of a melphalan and prednisolone combination because of financial problems. Among those patients who underwent chemotherapy or chemoimmunotherapy, 5 achieved complete response (CR), 2 achieved partial responses (PR), and the remaining 3 achieved stable disease. The response was not evaluated in 1 patient because of loss of clinical follow up. Glucocorticoid alone was used in 6 (21%) patients, 2 of whom achieved CR and 3 of whom experienced PR. The remaining patient could not be evaluated. The duration of glucocorticoid treatment was variable, ranging from 2 weeks with daily administration to 16 months with intermittent dosing. One patient received interferon-alpha at 3 million units per week for 15 weeks with prednisolone and achieved CR. Five patients were treated with excision, and 1 of them underwent postoperative radiation therapy. The other 5 patients (32%) were treated using a "watch and wait" strategy without systemic therapy. The patients who underwent surgery or surveillance did not initially have constitutional symptoms, except for 1 who presented with fever that resolved without treatment. All of the patients treated with local therapy had disease on a single side of the diaphragm. Two patients who were not administered specific therapy (i.e., watch and wait strategy) had diseases on both sides of the diaphragm. The overall response rates (CR+PR) in patients who underwent systemic therapy (i.e., cytotoxic chemotherapy, glucocorticoid, or interferon-α) was 72% (13/18).
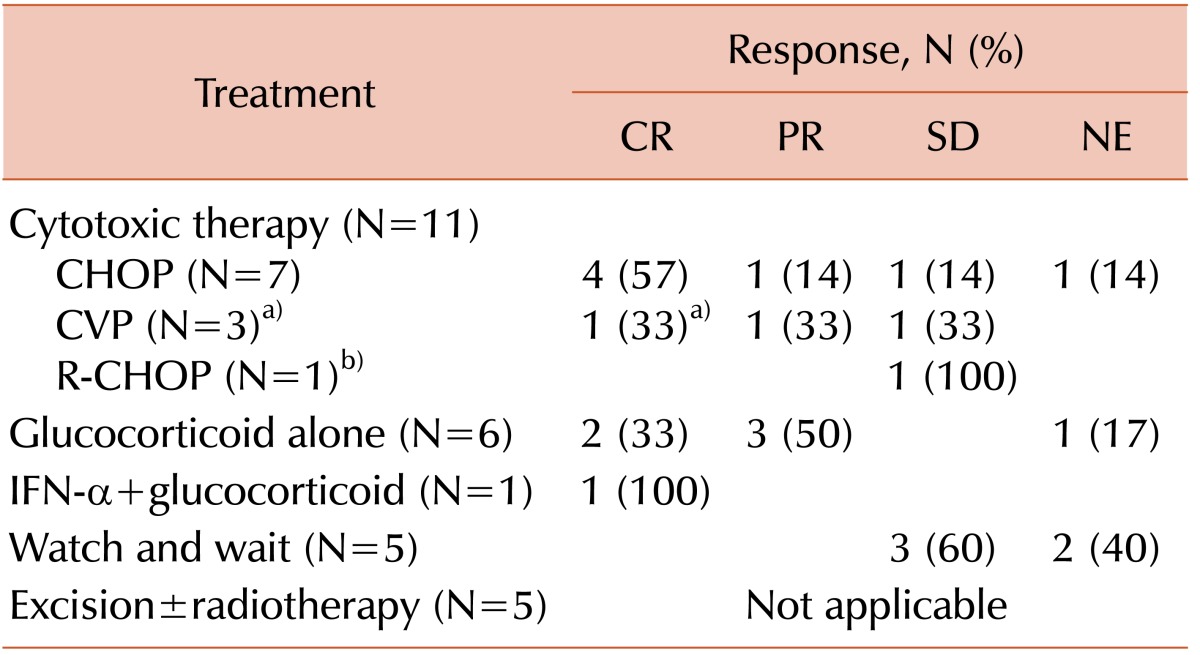
Table 3
Initial treatment and best response.
a)Rituximab was added from the second to sixth cycle in a patient with Evan's syndrome; this patient achieved CR.
b)A patient received 1 cycle of rituximab plus CHOP followed by melphalan and prednisolone.
Abbreviations: CHOP, cyclophosphamide, adriamycin, vincristine, prednisolone; CR, complete response; CVP, cyclophosphamide, vincristine, prednisolone; INF-α, interferon-alpha; NE, not evaluable; PD, progressive disease; PR, partial response; R-CHOP, rituximab plus CHOP; SD, stable disease.
![]()
At the time of analysis, 10 of the 28 patients had died. Two died from secondary lymphoma, each with T-cell lymphoma with leukemic manifestation and high-grade B-cell lymphoma. Three died from disease progression. The cause of death was unknown in 5 patients because of follow-up loss. Among those 10 dead patients, 7 had diseases on both sides of the diaphragm and 6 had clinical features related to extravascular fluid accumulation.
With a median follow-up of 67 months (range, 1-162 months), the 5-year OS rate of all patients was 77% (95% confidence interval [CI], 69-86); it was 100% in those who underwent local therapy and 70% (95% CI, 59-82) in those who were given systemic therapy. When we excluded 2 patients from the systemic therapy group given rituximab, the 5-year OS rate was 66% (95% CI, 54-79).
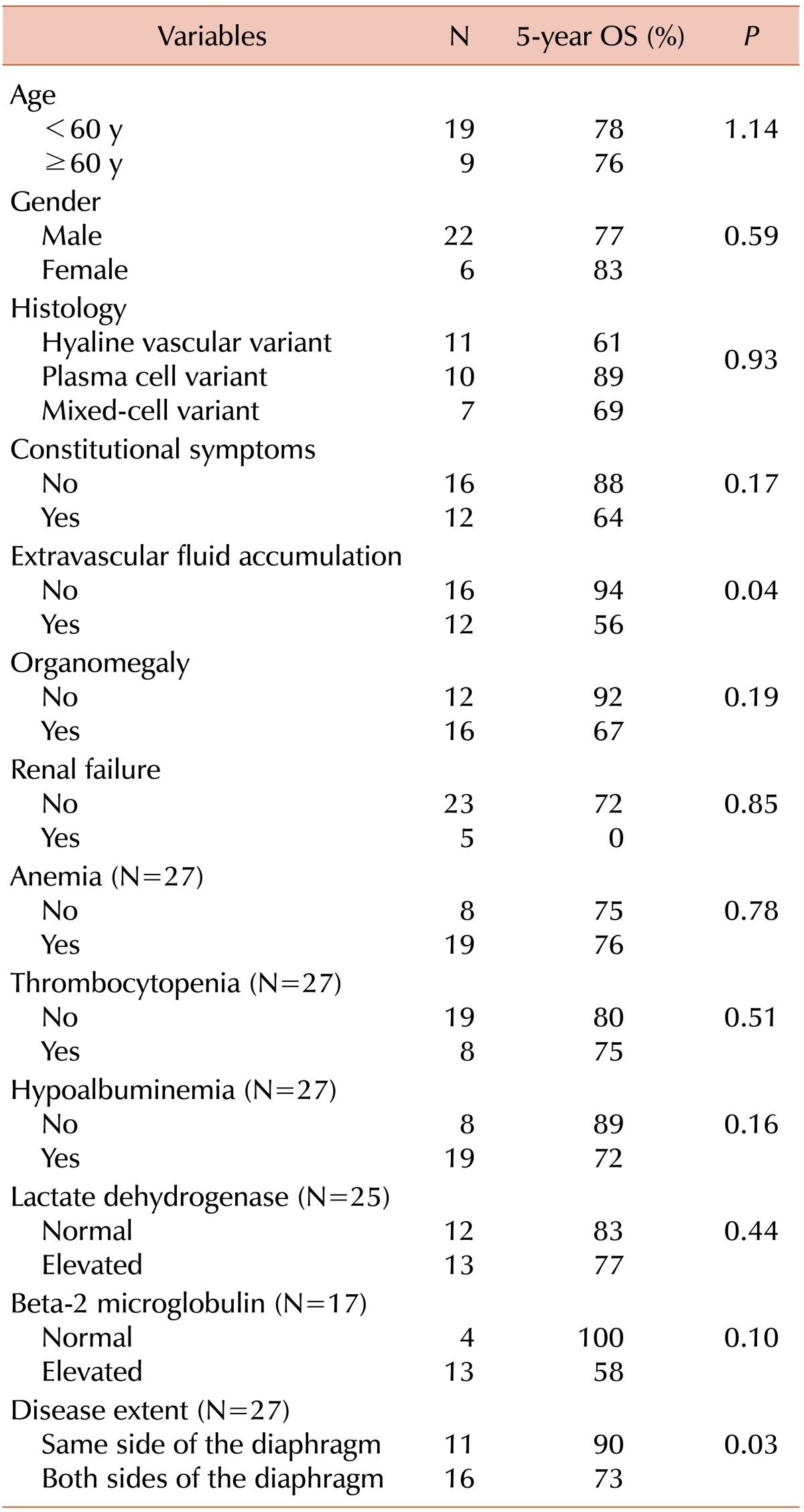
Univariate analyses for potential prognostic factors were performed (Table 4). Patients with extravascular fluid accumulation, such as peripheral edema, ascites, and/or pleural effusion (5-year OS rates 56% vs. 94%, P=0.04) and disseminated disease extent (5-year OS rates 73% vs. 91%, P=0.03) were associated with poor survival (Fig. 1). Elevation of β2 microglobulin was marginally associated with poor survival (5-year OS rates, 58% vs. 100%; P=0.10). Other clinicopathologic factors, including patient gender, age, histopathologic types, existence of organomegaly, hypoalbuminemia, anemia, thrombocytopenia, and elevated lactate dehydrogenase, were not significantly associated with their survival. HHV-8 positivity by either PCR or IHC also was not correlated with their survival (P=0.25). Because of the small sample size, multivariate analysis was not performed in this study.

Go to :
DISCUSSION
With the extensive heterogeneity of its clinical features, the natural courses and prognosis of multicentric CD have not been clarified. Because our study cohort was composed of recently diagnosed patients, they were relatively well evaluated with imaging and pathologic workup.
In our study focusing on HIV-negative multicentric CD patients, the patients' clinical characteristics were consistent with those seen in previous studies [11, 12]. However, our cohort had more patients with HVV. Previous studies have reported that multicentric CD histology was mainly a plasma cell variant (75-100%) [10, 11, 13], whereas other recent studies have shown a lower proportion of plasma cell variants (38-52%) with a greater percentage of HVV (28-33%) in multicentric CD regardless of HIV infection status [14, 15, 16]; this is consistent with our results. These findings suggest that HVV in multicentric CD is not that rare of a subtype, unlike what was observed in previous reports.
The 5-year OS rate of our cohort (77%) seems to be favorable compared with those of previous reports, in which the 5-year OS rates ranged from 50% to 65% in mostly HIV-negative multicentric CD cohorts [10, 15]. Although a role for rituximab has been suggested in HIV-positive multicentric CD patients [9, 17], its role in HIV-negative multicentric CD patients has not yet been established [18, 19]. Among our patients, only 2 received rituximab. One patient treated with 5 cycles of rituximab plus CVP has been disease-free for 6 years; another who received 1 cycle of rituximab plus CHOP followed by melphalan and prednisolone died of disease progression. Our data have limitations in evaluating rituximab's role in treating CD; this should be explored in prospective trials.
The extent of extravascular volume overload and disease were significant prognostic factors in this study. Elevated vascular-endothelial growth factor (VEGF) in tissue and sera is known to induce angiogenesis and vascular leakage, both of which were reported as possible pathomechanisms in multicentric CD. VEGF levels are also known to be increased by interleukin-6, which is a key cytokine in CD pathogenesis [20, 21]. Although we did not directly measure the levels of both VEGF and interleukin-6, we believe that extravascular fluid accumulation might be related to elevated VEGF levels, which in turn are possibly related to disease activity in multicentric CD-like POEMS syndromes [22]. Considering that CD is a type of lymphoproliferative disorder, it is not surprising that disease extent was a significant prognostic factor, although this concept has not previously been proposed in CD.
Our study has several limitations. This is a small-sized, single-center, retrospective analysis that included patients with heterogeneous treatments. Furthermore, with the statistical association between prognostic factors and lack of multivariate analysis, we cannot make robust conclusions. Because of the rarity of the disease, however, this is one of the largest studies on multicentric CD in the Asian population.
In conclusion, our data suggest that HVV in multicentric CD is not a rare subtype. Extravascular fluid accumulation and disseminated disease might be significant prognostic factors for multicentric CD, and their prognostic relevance requires validation in future studies.
Go to :
 XML Download
XML Download