

 PDF
PDF ePub
ePub Citation
Citation Print
Print


INTRODUCTION
Natural killer (NK) cells belong to the subpopulation of lymphocytes capable of recognizing and killing tumor cells as well as virus infected cells without pre-sensitization [1]. NK cells are characterized by the expression of CD56 and CD16 antigens, but do not express CD3 (a general T lymphocyte marker) or CD19 (a general B lymphocyte marker) [2]. CD56 is an adhesion glycoprotein commonly used to identify NK cells, although some NK cells express T cell markers CD3 and CD4 [3]. CD16 has been identified as low-affinity Fc receptors FcγRIII. Because CD16 is a marker of differentiation found on the surface of NK cells, it can be used to isolate populations of NK cells using magnetic activated cell sorting system [3].
NK cell activity is controlled by a delicate balance of signals derived from activating and inhibitory receptors. NKp30, NKp44, and NKp46 are important receptors for NK cell activation. In particular, NKp44 is a representative marker of IL-2-induced activated NK cell [4]. NKG2D and DNAM-1 are also important receptors for NK cell activation. Among these, NKG2D has generated considerable interest of late. In addition to NK cells, NKG2D is also expressed on αβT and γδT cells [5]. NK cells use NKG2D as a primary activation receptor and trigger killing activity. DNAM-1 belongs to the immunoglobulin superfamily and utilizes CD155 and CD112 as cell surface ligands [6]. The cytolysis activity of NK cells is controlled by inhibitory receptors. These receptors are bound to MHC class I molecules, which are generally expressed on most healthy cells, but are down regulated by virus infection or during cancer development. KIR and CD94/NKG2A are most studied inhibitory receptors in human body.
NK cells play a pivotal role in innate and acquired immune responses. Therefore, attempts to develop NK cells expansion methods have been made using cytokines, chemicals, and feeder cells [7, 8]. Cytokines play central roles in the modulation of NK cell activity. IL-2, IL-12, IL-15, IL-18, IL-21, and IFN-γ are classified as activation cytokines for cytotoxicity and proliferation of NK cells [9]. Among these cytokines, IL-2 can induce proliferation of NK cells and enhance cytotoxicity against a broad range of human tumor cells. In a previous study, IL-2 activated human naïve NK cells were cultured over a number of weeks for high levels of proliferation and cytotoxicity [10]. Feeder cell culture is one of the promising methods for expanding NK cells from blood-derived cells [10]. Blood-derived peripheral blood mononuclear cells (PBMCs), cord blood-derived mesenchymal stem cells, and lymphocyte-derived cancer cell lines have been reported as feeder cells [11, 12]. In these studies, feeder cells were used in combination with IL-2 or cytokine cocktails, because coculture with feeder cells without cytokines did not yield any NK precursor expansion at all.
Of all cancer cell lines, human myelogenous leukemia K562 and human Burkitt's lymphoma cell line Daudi have been used as feeder cells. The K562 feeder enhanced proliferation and cytotoxic activity of matured NK cells (CD16+/CD3-cells) in the presence of recombinant human IL-2 [13]. Daudi enhanced proliferation and differentiation of NK cells from bone marrow cells in the presence of recombinant human IL-2 [10]. However, these studies neither compare the feeder activity between cancer cells nor demonstrate the mechanisms of feeder cells. Therefore, the present study designed a test to evaluate different feeder cells' abilities to enhance NK cell expansion from PBMCs.
Go to : 
MATERIALS AND METHODS
Cell lines
Hep3B (human hepatocellular carcinoma), Jurkat (T cell leukemia cell line), K562 (human myelogenous leukemia cell line), MCF-7 (human breast cancer cell line), Raji (human Burkitt's lymphoma cell line), Ramos (human Burkitt's lymphoma cell line) were purchased from American Type Culture Collection (ATCC, Rockville, MD). These cell lines were cultured with 10% fetal bovine serum/RPMI 1640 (Hyclone Laboratories, Logan, UT) supplemented with penicillin (100 U/mL) / streptomycin (100 µg/mL) (Gibco BRL, Grand Island, NY).
Preparation of NK cells
Blood samples were obtained following acquisition of the study participants' written informed consent. Human PBMCs were isolated from healthy donors by density gradient centrifugation using Ficoll-Hypaque Plus (Amersham Biosciences, Piscataway, NJ) and then CD3+ lymphocyte was depleted by immunomagnetic beads selection using DynabeadsCD3 (Invitrogen, Dynal, Inc., Oslo, Norway), according to manufacturer's instructions. CD3+ depleted lymphocytes (CD3dep cells) (1×107 cells/mL) were mixed with IL-2 (100 IU/mL) and added to Ø90 tissue culture plate containing feeder cells treated with 20 µg/mL mitomycin-C (Sigma) for 30 min at 37℃ / 5% CO2. The culture medium was replaced with fresh culture medium containing IL-2 every three days. Cells were harvested and counted with trypan blue exclusion and analyzed using flow cytometry. The number of NK cells was calculated as follows: number of NK cells=number of final expanded CD3dep cellsx00D7;[final% of CD56+ and CD16+ cells double positive cells].
Phenotypic analysis
The expression of NK cell markers on CD3dep cells before and after expansions was analyzed by flow cytometry on a FACS Caliver (Becton-Dickinson, Franklin Lakes, NJ). In the analysis, 10,000 cells were scored. Fluorescein isothiocyanate (FITC)-conjugated anti-CD16 and anti-NKG2D were obtain form BD Pharmingen (San Diego, CA). Phycoerythrin (PE)-conjugated anti-CD56, allophycocyanin (APC)-conjugated anti-DNAM-1, Mouse IgG1 APC isotype control (Clone 11711), Mouse IgG1 PE control (Clone 11711) were obtain form R&D system (Minneapolis, MN, USA).
Intracellular staining
Expanded NK cell clones were stained for intracellular perforin (PE-conjugated antibody from BD Pharmingen) and granzyme B (FITC-conjugated antibody from BD Pharmingen) after fixation (4% paraformaldehyde) and permeabilization (0.1% saponin, 1% BSA, 0.05% Tween-20 in phosphate buffer saline). They were evaluated on flow cytometric analysis. Data were presented according to mean fluorescence intensity (MFI).
Cytotoxicity assay
NK cell cytotoxicity towards cancer cell lines was measured using lactate dehydrogenase (LDH) release assay from Takara (Japan) according to manufacturer's protocol. Using a 3:1 effector cell-to-target cell ratio, 1.8×105 NK cells were plated in a round bottom 96-well plate. Cancer cell lines were added to the wells, and the plate was then incubated at 37℃ / 5% CO2. After 4 and 6 hours, cells were pelleted with centrifugation at 250 g for 10 min. The 100 µL of supernatant was transferred to a new 96-well plate and 100 µL of LDH substrate mix was added. After 30 min incubation in the room temperature, the absorbance was measured at 490 nm. Percentage of cytotoxicity was calculated as follows: cytotoxicity (%)=[(effector and target cell mix-effector cell control)-low control]/(high control-low control)×100, where effector cell control: culture supernatants of NK cells only; low control: culture supernatants of cancer cells only; and high control: culture supernatants of cancer cells after lysis with Triton X-100.
Statistical analysis
The data were analyzed using the Student's t-test to determine their statistical significance. The values were given as means±standard deviations (SD).
Go to :
RESULTS
Effects of feeder cells on NK cell expansion
In initial experiments, CD3dep PBMC cells were cocultured with mitomycin C inactivated feeder cells (PBMC, K562 and Jurkat) and IL-2 (100 IU/mL) for 15 days, at a CD3dep PBMC cells-to-feeder cells ratio of 3:1, and the total number of live cells was counted on Days 0, 3, 5, 7, 9, 11, 13, and 15 using a hemocytometer. As shown in Fig. 1, the culture with IL-2 in the absence of feeder cells exhibited a significantly lower growth than the cultures with feeder cells. Coculture with feeder cells without IL-2 did not yield any cell expansion (data not shown). Coculture using different CD3dep PBMC cells-to-feeder cells ratios showed inferior expansion (data not shown). The highest cell expansion was observed in IL-2 plus K562-feeder cultures (3.15±0.303-fold increase on Day 9). However, total cell expansion was not observed after stimulation with IL-2 plus PBMC feeder cells or Jurkat feeder cells.
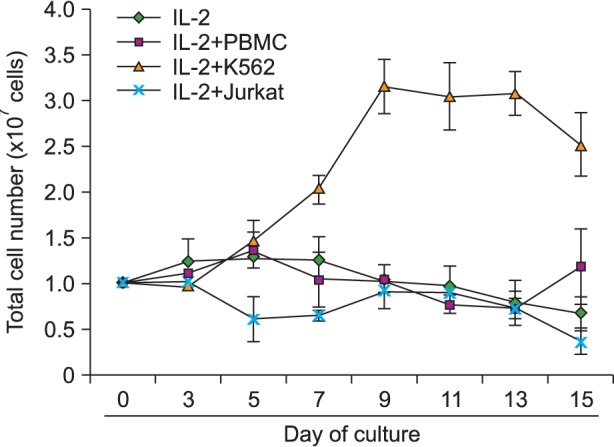 | Fig. 1Comparison of total PBMC expansion with and without feeder cells. CD3dep PBMC cells were cocultured with IL-2 and with feeder cells (PBMC, K562, and Jurkat) for 15 days. The total numbers of cells were counted on Days 0, 3, 5, 7, 9, 11, 13, and 15. The data from four independent experiments performed in triplicate are expressed as mean±SD.
|
CD3dep PBMC cell cultures with or without feeder cells were examined for distribution of CD56+/CD16+ double-positive NK cells and CD3+ T cells. As shown in Fig. 2A, the culture of CD3dep PBMC with IL-2 and K562 combination showed a substantial increase of CD56+/CD16+ double-positive cells. Although somewhat less, an increase in CD56+/CD16+ double-positive NK cells was also observed in IL-2 plus PBMC and IL-2 plus Jurkat-stimulated CD3dep PBMC cell cultures. These results were in contrast to the expansion of CD3+ T cells because the PBMC-stimulated CD3dep PBMC culture showed a substantial increase of CD3+ cells, whereas the K562-stimulated CD3dep PBMC culture showed a slight increase of CD3+ cells (Fig. 2B).
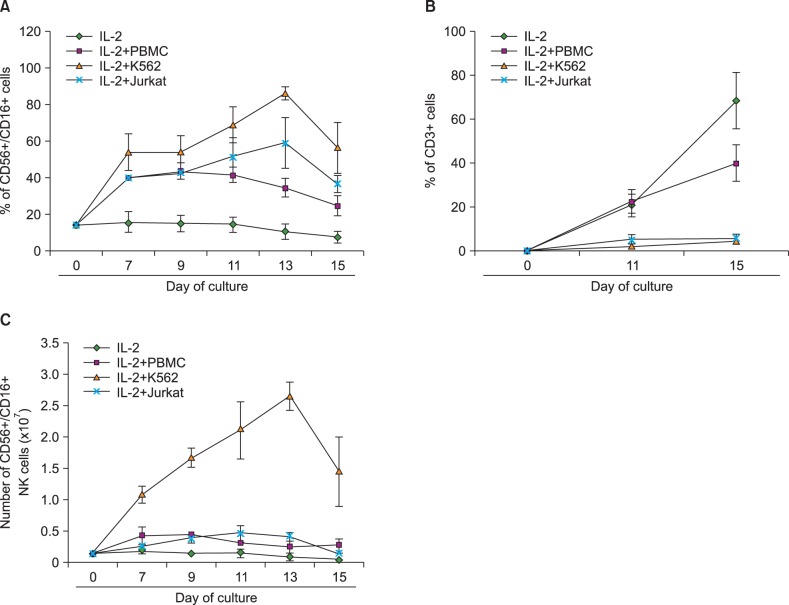 | Fig. 2Optimization of NK cell expansion. CD3dep PBMCs were cultured as described in Fig. 1 and the percentages of CD56+/CD16+ double-positive NK cells. (A) and CD3+ T cells. (B) were determined. The number of CD56+/CD16+ double-positive NK cells was counted using flow cytometry. (C) The data from three independent experiments performed in triplicate are expressed as mean±SD.
|
Based on Fig. 1 and 2A, the number of CD56+/CD16+ double-positive NK cells was calculated (Fig. 2C). As expected, the stimulation of CD3dep PBMC with IL-2 and K562 resulted in the highest expansion of NK cells (19±3.33-fold). Some expansion of NK cells was also observed in the PBMC-feeder (maximum 2.29±0.37 on Day 11) and Jurkat-feeder (maximum 3.44±1.23 on Day 11) cultures; however, the effects were 5-8 times less than what was observed in K562-feeder cultures. There were no major changes in the culture with IL-2 in the absence of feeder cells.
Effects of feeder cells on the generation of NK cell cytotoxicity
Cytotoxic activity of the expanded CD3dep PBMC with IL-2 and feeder cells was measured on Day 12 against K562, Jurkat, Hep3B, Raji, MCF-7, and Ramos. As shown in Fig. 3, the highest cytotoxicity against K562, Jurkat, Raji, MCF-7, and Ramos was observed in the K562-feeder-expanded NK cells (K562-NK cells). NK cells expanded with PBMC (PBMC-NK cells) or Jurkat feeder (Jurkat-NK cells) were also effective in generating cytotoxicity against the same targets, but these activities were less prominent. These results indicate that K562 is a more beneficial feeder than PBMC and Jurkat for enhancing NK cell expansion and activation. Interestingly, all of the expanded CD3dep PBMC with K562, PBMC, and Jurkat feeder cells showed relatively low cytotoxicity against Hep3B compared with other cancer cell lines. We also observed changes in the cytotoxicity of K562-NK cells during expansion periods. As shown in Fig. 3B, cytotoxicity of K562-NK cells exhibited peaks against Hep3B on Day 11 and against Jurkat on Day 13, followed by decreases. However, K562 showed a steady susceptibility to K562-NK cells. These results were expected, because CD56+/CD16+ double-positive NK cells were observed with the highest frequency on Day 13.
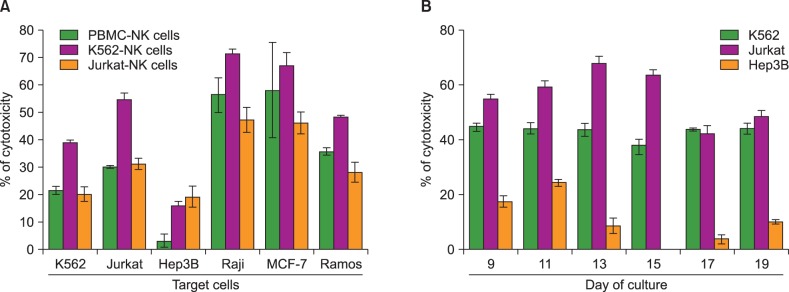 | Fig. 3Comparison of cytotoxic properties of NK cells expanded with PBMC, K562, and Jurkat feeder cells. Expanded NK cells were tested for cytotoxicity against K562, Jurkat, Hep3B, Raji, MCF-7, and Ramos. (A) Cytotoxic activity of K562-NK cells was measured according to culture duration. (B) Cancer cell lines (6×104) and NK cells (1.8×105) were added on to the plates and incubated for 4 hours at 37℃. NK cell cytotoxicity towards cancer cell lines was measured using LDH release assay. The data from four independent experiments performed in triplicate are expressed as mean±SD.
|
To demonstrate the possibility of K562-NK cells as a cell therapeutic material, we compared the cytotoxic activity between K562-NK cells and naïve NK cells. For precise verification, cytotoxicity assays were performed with two different incubation times (4 and 6 hours). As shown in Fig. 4, K562-NK cells exhibited a more potent cytotoxicity than naïve NK cells regardless of the type of cancer cells and incubation time. In particular, more prominent levels of cytotoxicity against Hep3B, Raji, and Ramos were observed in K562-NK cells than in naïve NK cells.
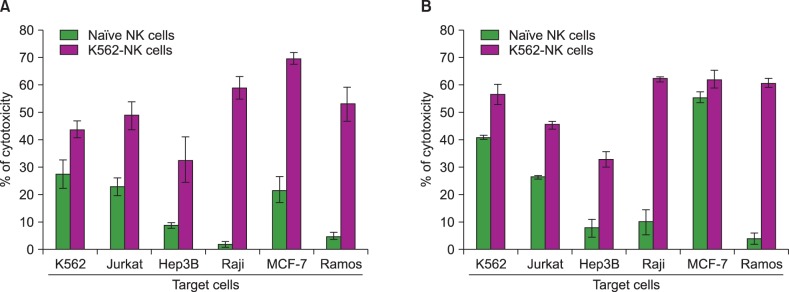 | Fig. 4Comparison of cytotoxic properties of naïve NK cells and K562-feeder expanded NK cells. Cytotoxic activities of naïve and K562-NK cells were compared using LDH assay. Cancer cell lines and NK cells were added onto the plates and incubated for 4 hours (A) and 6 hours (B) at 37℃. The data from three independent experiments performed in triplicate are expressed as mean±SD.
|
Expression of surface antigen on expanded NK cells
To investigate why K562-NK cells showed more potent cytotoxicity than other NK cells, we analyzed the expression of NK cell activation receptors, NKG2D, and DNAM-1, on NK cells using flow cytometry. As shown in Fig. 5, K562-NK cells remarkably expressed both NKG2D and DNAM-1. Almost 95.3% of K562-NK cells expressed NKG2D and DNAM-1 double-positive NK cells. In addition, 99.6% of the cells expressed at least one of the two receptors. Although less than K562-NK cells, a substantially high percentage of Jurkat-NK cells (62.8%) expressed both NKG2D and DNAM-1. A remarkable finding was that Jurkat-NK cells expressed a relatively low percentage of DNAM-1 compare to K562-NK cells. 85.4% of naïve NK cells expressed DNAM-1, whereas just 0.8% expressed NKG2D.
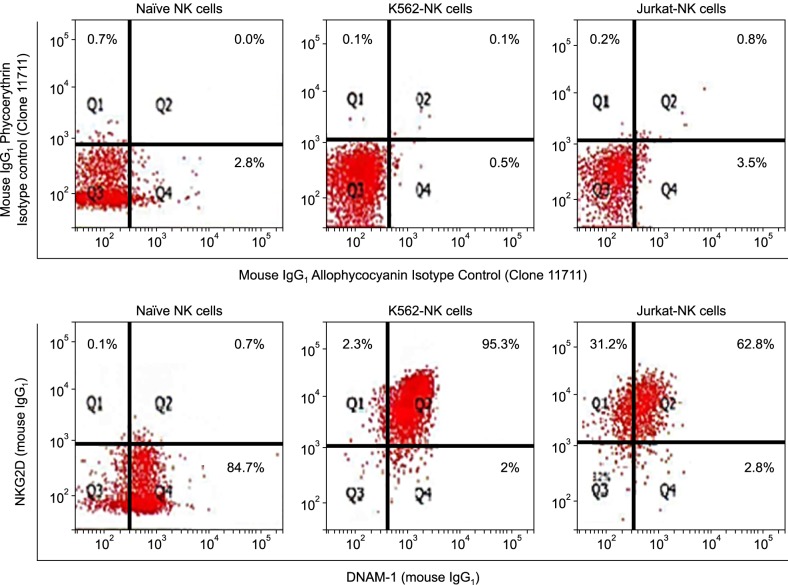
Production of cytotoxic granule in expanded NK cells
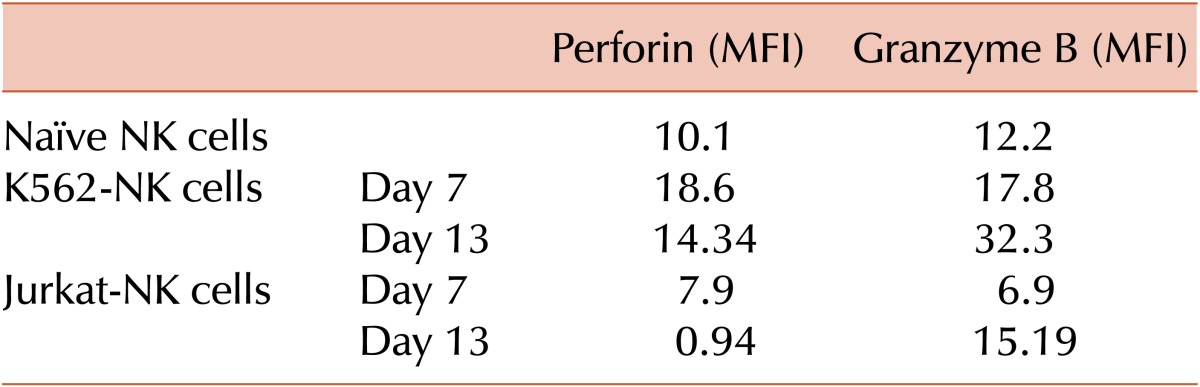
NK cell cytotoxicity is a complicate process that requires adhesion to target cells, synapse formation, and cytotoxic granule production, as well as exocytosis. To investigate the mechanisms for feeder-induced NK cell cytotoxicity, productions of cytotoxic granule, perforin, and granzyme B were analyzed using intracellular immunofluorescence staining and flow cytometry analysis. naïve NK cells were used as a positive control after 24-hour stimulation with IL-2. Expanded feeder-NK cells were harvested on Days 7 and 13, permeabilized with saponin, and then stained with anti-perforin-PE and anti-granzyme B-FITC monoclonal antibody. As shown in Table 1, K562-NK cells exhibited more than two-fold production of both cytotoxic granules compared with Jurkat-NK cells, producing more perforin and granzyme B than naïve NK cells.
Go to :
DISCUSSION
Recently, many researchers and clinicians have tried cell therapy to remove tumors from cancer patients. In the process, many types of immune cells, such as dendritic cells, cytotoxic T cells, and NK cells, were examined as potential effector cells for cancer therapy. Among them, NK cells have been recognized as the strongest and most important candidate. Dendritic cells and cytotoxic T cells can remove cancer cells after recognizing cancer antigens. In contrast, NK cells constantly survey surrounding tissues and remove newly generated cancer cells, independent of cancer antigen recognition. Although many kinds of autologous and allogenic NK cells have been applied in cancer therapy [1], clinical application has been somewhat limited because it is difficult to prepare a sufficient number of NK cells. Therefore, ex vivo NK cell expansion is the most important step for developing NK cell therapy.
In earlier studies, many researchers have tried various methods of ex vivo NK cell expansion to develop NK cell therapeutics [14]. PBMCs have been used as a general source of NK cells for clinical application [14]. PBMCs are composed of many kinds of mature and immature leukocyte, and NK cells and NK progenitor cells are also types of PBMCs. Therefore, whole PBMCs can be used as a source of NK cell expansion. These results are partially consistent with our results obtained using K562 feeder cells. In our experiment, we used CD3dep PBMCs and achieved a 19-fold increase in NK cells after 13 days. In the process, CD3dep PBMCs were used as a general source of NK cells [15]. CD3dep PBMCs were enriched with CD56+ cells to increase the number of activated NK cells [15]. However, a few reports have claimed that applying CD3dep PBMCs and cancer feeder cells simultaneously. Furthermore, several papers have compared feeder cell activities for NK cell expansion.
In this study, we compared feeder activities of three different cells-PBMC, K562, and Jurkat. K562 and Jurkat are types of human leukemia cell lines and frequently used as positive controls to indicate cytotoxic activity of NK cells. Therefore, K562 and Jurkat were selected as candidate feeders for expanding the NK cell population. K562 weakly expresses proteins that inhibit NK cell cytotoxicity, such as MHC class I molecules, because K562 cannot send inhibitory signals to NK cells. In turn, K562 is easily attacked by NK cells. In previous studies, cancer cells (Wilms tumor cell line) [16], B lymphoblastoid cell lines [2], malignant melanoma cell lines [17] and naïve human monocyte [18] that weakly express MHC-class molecules were used as feeder cells to expand NK cells. Genetically modified or ligand transfected K562 was also used to increase the number of activated NK cells. Indeed, the modified K562 cells expressing 4-1BB ligand and IL-15 enhanced NK cell expansion almost 100-fold [19]. Genetically modified K562-based antigen presenting cells expressing membrane-bound IL-21 promoted NK cell expansion almost 47,000-fold [20]. On the other hand, Jurkat expresses a high level of MHC class I molecules but is also regarded as an NK-susceptible target [21]. These results contradict the general theory. In our previous study [22], NK cells showed the most potent cytolytic effect against Jurkat compared to other cancer cell lines, such as MCF-7, Raji, Ramos, and even K562. We found that Jurkat highly expresses activation molecules and NKG2D ligands, the results of which are easily exposed to NK cells. Therefore, we believe that the expansion capacity of NK cells is influenced by the expression levels of MHC class I cells on the surface of feeder cells, but that would not rule out other reasons.
In previous studies, the various attempts were made to stimulate NK cell expansion with irradiated autologous PBMCs. Lim et al. [23] showed the simple and efficient NK cells expansion method with irradiated autologous PBMCs in the presence of OKT3 and IL-2. Ahn et al. [24] also developed a NK cell expansion method, using irradiated and activated autologous PBMCs. The similarity of the two papers is that autologous PBMCs and variety of additives including IL-2 were used. However, our results showed that PBMC induced very weak expansion of NK cells; this effect was much less than what was observed in previous results [23, 24]. This disparity may be due to the differences of feeder cell treatment (mitomycin C versus irradiation) and sources (allogeneic versus autologous).
NK cells can identify and kill cancer cells, resulting in phenotypic changes when a tissue with normal growth pattern turns into a malignant tumor. Malignant cells reduce the level of MHC class I molecules, while elevating the level of NK cell-activating ligands, including those that bind to NKG2D and DNAM-1. Previous studies have demonstrated that NKG2D and DNAM-1 play important roles in killing tumor cells [25]. NK cells express NKG2D and DNAM-1 that recognize ULBPs and MICA/B, or CD155 and Nectin-2 (CD112), respectively. These cellular ligands are upregulated or newly expressed in the course of neoplastic transformation.
NKG2D is regarded as a primary activating receptor of NK cells, and its ligands are stress-induced proteins such as MICA, MICB, ULBP1, ULBP2, ULBP3, and ULBP4 [26]. NKG2D ligands activate NK cells through phosphatidyl inositol-3 kinase, resulting in increased perforin-dependent lysis of ligand-expressing target cells [27]. The importance of NKG2D has been demonstrated in animal experiments. Guerra et al. [25] showed that NKG2D-deficient mice were highly susceptible to primary tumorigenesis in vivo, confirming the important role of NKG2D in tumor immune surveillance. DNAM-1 is also regarded as an important co-activating receptor in cancer immunity. DNAM-1 participates in cancer cell recognition especially in neuroblastoma [28], ovarian carcinoma [29]. Mouse DNAM-1 partially contributes to NK cell-mediated lymphoma rejection [30], but its relevance for NK cell immunity to melanoma is unknown. Our experiments showed that K562-NK cells express more NKG2D and DNAM-1 receptors than both Jurkat-NK and naïve NK cells. This result is consistent with the theory that the expression of NKG2D and DNAM-1 receptors is associated with the cytotoxic activity of NK cells.
In summary, our findings suggest that K562 are more efficient feeder cells than Jurkat or PBMCs. K562 feeder cells expanded NK cells by almost 20-fold and showed powerful cytotoxic activity against cancer cells. We herein propose an intriguing approach for a design of NK cell expansion for development of NK cell cancer therapeutics.
Go to :
 XML Download
XML Download