

 PDF
PDF ePub
ePub Citation
Citation Print
Print


INTRODUCTION
Chronic myeloid leukemia (CML) is a myeloproliferative neoplasm with clonal hyperproliferation of myeloid cells in the bone marrow. Clinical findings in chronic-phase CML include fatigue, decreased appetite, and splenomegaly, and hematological findings include leukocytosis, thrombocytosis, neutrophilia, and decreased leukocyte alkaline phosphatase (LAP) scores [1].
Isolated thrombocytosis is assumed to be essential thrombocythemia (ET) rather than chronic-phase CML in most cases. According to the 2008 World Health Organization diagnostic criteria, a diagnosis of ET is routinely made, if platelet counts exceed 450×109/L with proliferation of megakaryocytes in the bone marrow and JAK2 V617F mutation, excluding any evidence of other myeloproliferative neoplasms [2]. Therefore, cases of isolated thrombocytosis with Philadelphia chromosome or bcr/abl rearrangement, which are diagnostic markers for CML, should not be diagnosed as ET. In previous studies, however, patients have been diagnosed with Philadelphia chromosome-positive ET [3, 4]. Other contradictory results described them as variants of CML [5, 6].
We experienced a case of isolated thrombocytosis, initially suggestive of ET, positive for the Philadelphia chromosome and bcr/abl rearrangement. The patient was eventually diagnosed with chronic-phase CML, and was successfully treated with imatinib. Here, we report our case and include a literature review.
Go to : 
CASE REPORT
A 21-year-old woman presented to the outpatient clinic with lower abdominal pain. Seven days before presentation, she had a ruptured corpus luteal cyst, which was detected on abdominal computed tomography (CT) at another clinic. Her initial platelet count was estimated to be 3,777×109/L at our clinic. Because thrombocytosis appeared to be secondary to bleeding, the patient's blood cell counts were only monitored during her clinical course. However, we decided to perform further evaluation because of thrombocytosis persisted for 2 weeks with no decrease in the platelet count. Her medical history was unremarkable, and she had no family history of hematologic disease or genetic disorders. Her vital signs were normal at admission. Except for mild lower abdominal tenderness, the patient had no other positive findings on physical examination. Complete blood count (CBC) revealed a hemoglobin level of 10.1 g/dL, hematocrit level of 30.7%, white blood cell (WBC) count of 10×109/µL (differential count: neutrophils 63%, lymphocytes 33%, eosinophils 1%, basophils 3%, and monocytes 0%), and platelet count of 3,294×109/L. A serum biochemistry panel showed the following: total protein, 7.2 g/dL; albumin, 3.9 g/dL; total bilirubin, 1.2 mg/dL; aspartate aminotransferase, 11 IU/L; alanine aminotransferase, 13 IU/L; blood urea nitrogen, 6 mg/dL; creatinine, 0.6 mg/dL; lactic dehydrogenase, 410 IU/L; and C-reactive protein, 2.0 mg/dL. A peripheral blood smear showed thrombocytosis. In addition, serum iron level was 73 µg/dL, total iron binding capacity was 267 µg/dL, and ferritin level was 206.5 ng/mL. The patient had an LAP score of 127 points, which was within the normal range. Abdominal and pelvic CT showed a small amount of hemoperitoneum resulting from the previous ruptured ovarian cyst. Bone marrow aspiration and biopsy revealed a high number of megakaryocytes, but no cells undergoing malignant transformation (Fig. 1, Fig. 2). A cytogenetic abnormality was detected with the karyotype 46,XX,t(9;22)(q34;q11.2) on bone marrow. We also observed a bcr/abl rearrangement in the bone marrow using reverse transcriptase PCR, which also showed amplified products from the b3a2 mRNA deletion in the major bcr gene. Results were negative for the JAK2 V617F mutation. Because the patient had isolated thrombocytosis (3,294×109/L), she was tentatively diagnosed with ET before the results of the cytogenetic and molecular studies were available, even if results for the JAK2 V617F mutation were unknown. Hydroxyurea was administered to the patient at a dose of 2,000 mg/day for 14 days to lower her platelet count. A follow-up CBC showed persistent thrombocytosis, platelet counts of 2,206×109/L, and leukocytopenia (1.1×109/L). We stopped hydroxyurea and identified the Philadelphia chromosome and bcr/abl rearrangement, but no JAK2 V617F mutation. This led to the final diagnosis of chronic-phase CML, for which the patient received imatinib. In the 6 days following the treatment with imatinib, the patient's platelet count normalized to 438×109/L. She is currently followed up to confirm complete molecular response against bcr/abl rearrangement. In the 3 months after treatment with imatinib, a major molecular response (3-log reduction of transcript levels) was observed.
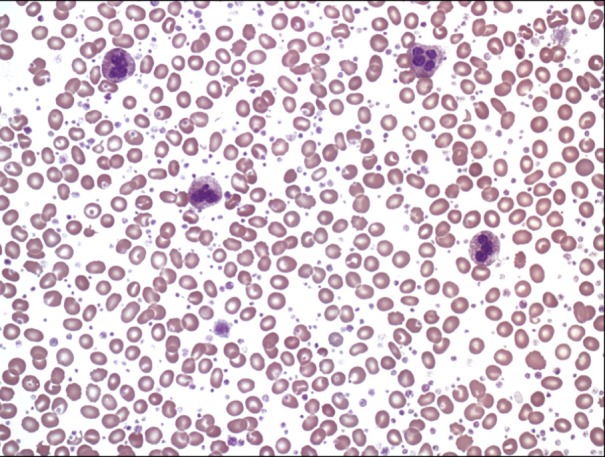
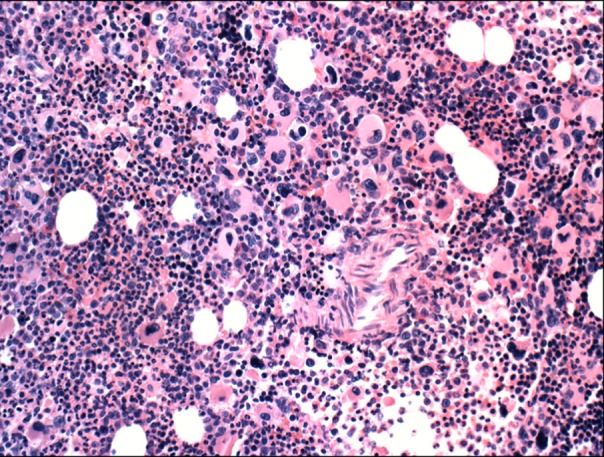
Go to :
DISCUSSION
Recently, a therapeutic advance in the treatment of CML was achieved with the advent of tyrosine kinase inhibitors for the bcr/abl protein. The long-term survival rate of patients with CML has improved to 89% [7]. To determine a distinctive diagnosis, we should distinguish CML from ET, which might have similar laboratory findings at initial diagnosis because of the different therapeutic approaches and clinical outcomes between the two diseases.
If severe thrombocytosis and hyperproliferation of megakaryocytes in the bone marrow occurs in the absence of splenomegaly, it is difficult to make a differential diagnosis between CML and ET without cytogenetic and molecular studies. Although the JAK2 V617F mutation is observed in approximately 50% of ET patients, JAK2 V617F mutation does not indicate a definitive diagnostic of ET, because ET should be confirmed with an exclusive diagnostic process. Therefore, the Philadelphia chromosome or bcr/abl rearrangement is a significant diagnostic tool for distinguishing CML from ET under these hematologically abnormal conditions.
A previous study showed mild basophilia and a Philadelphia chromosome in 1 of 121 patients with ET [8]. This patient had a Philadelphia chromosome and isolated thrombocytosis that progressed to the accelerated phase, which is similar to the clinical course observed in a patient with CML. Patients with a Philadelphia chromosome and isolated thrombocytosis considered a variant of CML or an initial presentation of CML in the chronic phase were refractory to hydroxyurea, but showed improvement in symptoms and laboratory values after administration of imatinib [6].
Imatinib mesylate (Gleevec) is a bcr-abl tyrosine kinase inhibitor and the primary treatment agent for patients with CML. By binding to bcr-abl protein tyrosine kinase and inhibiting the bcr/abl pathway, imatinib reduces the proliferation of bcr/abl-positive CML cells [9]. In patients initially diagnosed with chronic-phase CML, the rate of complete cytogenetic remission has increased to 76.2% in the imatinib era [10]. In addition, the overall survival rate significantly improved after treatment with imatinib in early-phase CML [7]. Therefore, imatinib is currently approved as a standard initial treatment for patients with chronic-phase CML.
In conclusion, our case indicates that CML could not be completely excluded until the presence of the Philadelphia chromosome or bcr/abl rearrangement was confirmed, which would be clinically sufficient to diagnose CML.
Go to :
 XML Download
XML Download