

 PDF
PDF ePub
ePub Citation
Citation Print
Print


INTRODUCTION
Hemophagocytic lymphohistiocytosis (HLH) is a systemic inflammatory disorder characterized by uncontrolled histiocytic proliferation, hemophagocytosis, macrophage activation, and up-regulation of inflammatory cytokines [1, 2]. Macrophage activation syndrome (MAS) is a subtype of HLH, recently classified as secondary or reactive HLH (RHLH) [3]. RHLH can occur secondary to underlying systemic diseases such as infection, immunodeficiency, or malignancy and to autoimmune diseases such as systemic-onset juvenile idiopathic arthritis [4], or Kawasaki disease (KD) [1, 5-11].
Kawasaki disease causes idiopathic panarteritis in infants and young children, predominantly affecting medium-sized vessels such as the coronary arteries. There are no specific laboratory tests for KD; therefore clinical criteria are used to diagnose it. The clinical criteria include prolonged fever (for more than 5 days), and at least 4 of the following signs: bilateral non-purulent conjunctivitis, cervical lymphadenopathy, polymorphous skin rash, oral or lip erythema, and edema or desquamation of the palms and finger tips [12]. It is well known that in young infants, KD may manifest with incomplete clinical signs [13]. A differential diagnosis between HLH and KD can be very difficult, as they are characterized by similar signs and symptoms. This is particularly so in cases of recurrent KD, with patients typically returning to the hospital with prolonged fever.
To date, there have been no clinical studies investigating the clinical spectrum of HLH-KD. Therefore, our goal was to estimate the incidence, clinical characteristics, and outcomes of HLH-KD in a nationwide survey of all members of the Korean Society of Pediatric Hematology-Oncology (KSPHO). Another goal was to investigate the clinical features that differentiate HLH from recurrent or refractory KD, to enable early diagnosis.
MATERIALS AND METHODS
We surveyed all members in the 21 institutions of the KSPHO between January 2012 and December 2012. The study protocol was approved by the Clinical Trials Committee of the KSPHO and by the Institutional Review Board of Hanyang University Hospital. Patients were eligible for this retrospective, multicenter study when their clinical and laboratory findings fulfilled the recognized diagnostic criteria for HLH and KD, respectively [1, 14].
We analyzed and compared the incidence of HLH-KD with the historical incidences of HLH and KD in Korea. We included in this comparison the clinical characteristics of HLH-KD, namely the interval between KD and HLH, the clinical and laboratory findings, treatment responses, and the outcomes. The software package SAS (version 9.2) was used to compare the clinical and laboratory findings with historical data from children diagnosed with HLH. The Kaplan-Meier method was used to estimate survival rates.
RESULTS
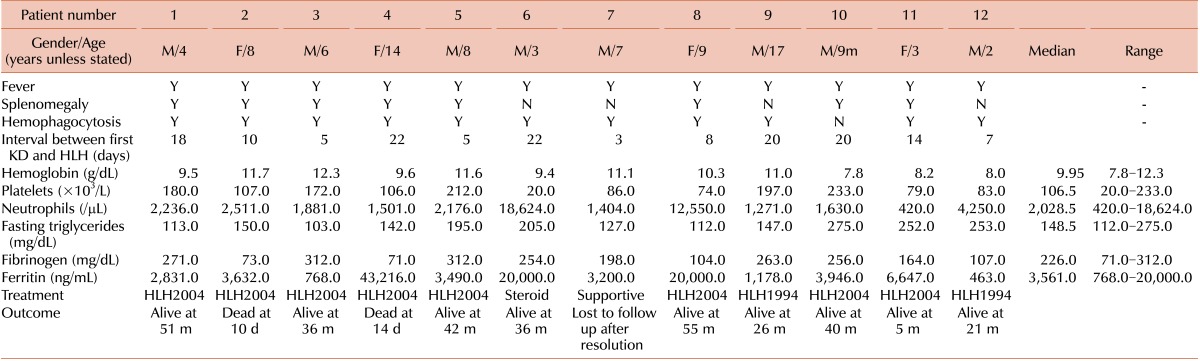
Twelve patients with HLH-KD, including 5 previously reported in Korea [11], who were diagnosed between April 2001 and December 2012 were recruited from 4 institutions. Although 4 cases did not strictly meet the diagnostic criteria of HLH-2004 [10], we decided to include these cases in this analysis for the following reasons: first, tests for NK cell activity and soluble CD25 level were not available in most institutions in Korea, and second, the physicians decided to treat their patients for HLH based upon strong evidence of hemophagocytosis as well as clinical and laboratory findings. The median patient age was 6.5 years (range, 9 months-14.7 years). Eight of the patients were male and 4 were female. The initial manifestations diagnosed as KD and accompanied by fever were as follows: polymorphous skin rash (in 11/12 patients), oral or lip erythema (6/12), bilateral non-purulent conjunctivitis (10/12), cervical lymphadenopathy (9/12), and edema or desquamation of the palms and finger tips (4/12). Coronary artery involvement during the first episode of KD was observed in 3 patients (2 on the right side, 1 on the left side). All the patients were treated with intravenous immunoglobulin (IVIG) with aspirin.
The median interval between the first episode of KD and the second visit with recurrent fever was 12 days (3-22 days). The clinical presentation at the second visit shared by all the patients was fever; associated clinical and laboratory findings included splenomegaly (in 8/12 patients) and hemophagocytosis in the bone marrow, spleen, or lymph nodes (11/12) (Table 1). The mean values of the laboratory tests were as follows: hemoglobin 9.95 g/dL (range, 7.8-12.3), neutrophils 2,028.5/µL (range, 420-18,624), platelets 106.5×103/µL (range, 20-233×103), serum triglycerides 148.5 mg/dL (range, 112-275), serum fibrinogen 226 mg/dL (range, 71-312), serum ferritin 3,561 ng/mL (range, 768-20,000), AST 108.5 U/L (range, 33-1,095), and ALT 106.5 U/L (range, 11-1,410). The data for NK cell activity were not available in all cases, and only 1 case was checked for soluble CD25 (5,869 U/mL). Genetic studies were performed in 2 of the 12 cases, which did not reveal any genetic mutations.
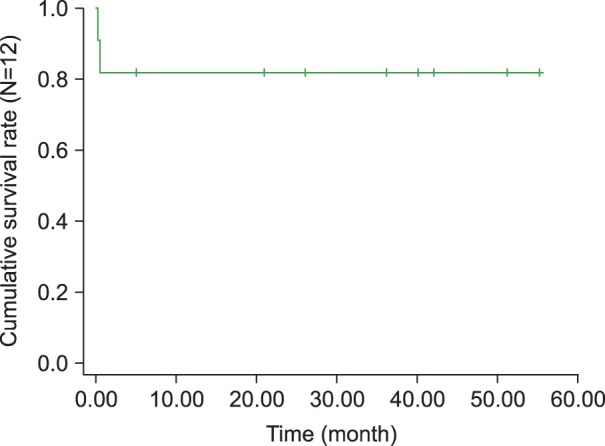
Two of the 12 children were initially treated with IVIG for recurrent KD when they presented again at the hospital with recurrent fever. Eventually, 8 of the 12 children were treated according to the HLH protocol 2004 and 2 according to the HLH protocol 1994. The remaining 2 cases received only supportive treatment including short-term steroids and/or antibiotics for 5 days and recovered. Two of the patients died of combined infections with disseminated intravascular coagulation and pneumonia at days 10 and 14 after chemotherapy, respectively, 1 resolved during a 10-day admission and was subsequently lost to follow-up, and 9 remain alive. The overall survival rate at 4 years was 81.1% with a median follow up of 45.1 months (Fig. 1).
DISCUSSION
HLH has 2 distinct etiological factors. The primary (or familial) form has been linked to inherited chromosomal factors, and specifically to mutations in PRF1 and MUNC13-4 [14]. The secondary (or reactive) form arises as a result of strong immune activation. Factors associated with secondary HLH include infection, malignancy, and other conditions that activate the immune system, such as KD [1, 2, 5-11].
The most frequently recognized cause of secondary HLH is the Epstein-Barr virus (EBV), responsible for 45.5-69.6% of cases of secondary HLH [15]. The incidence of HLH-KD among HLH patients is not known, as worldwide only 15 cases have been reported to date. The incidence of 23.8% reported by Kim et al. was based on data from a very small sample recruited at a single center [11]. Based on the recently enrolled data of the Histiocytosis Working Party of the Korean Society of Hematology, the incidence of secondary HLH due to KD among HLH patients in Korea would be estimated as 4.7% (12/253). Because the clinical manifestations of HLH are similar to those of KD, in particular to those of its recurrent form, differential diagnosis is very difficult. Since the 1990s, a nationwide epidemiological study has been conducted every 3 years in Korea to determine the epidemiological patterns and incidence of KD. In a recent report (2003-2005), the annual incidence of KD was 105.0/100,000 children <5 years of age, which was the highest reported in the world after Japan, and the recurrence rate was 2% [16]. Another study reported the recurrence rate as 2.3% (14/561), and stated that their 14 patients typically had laboratory results similar to the first episode of KD [17].
Early recognition and treatment of HLH is imperative to avoid fatal outcomes in severe cases [18]. Cummings et al. also concluded that testing for evidence of HLH in patients with recurrent KD (e.g. serum ferritin and triglyceride levels) could aid early diagnosis and treatment [9]. The mean time of KD recurrence from the first episode has been reported as 17.9 months (range, 1-60 months) [17], while in our study, the time to HLH onset following the first episode of KD was only 13.3 days (range, 3-22 days), suggesting a potential early diagnostic clue of HLH following KD. In only 3 of the 10 cases reported previously [1, 5-10], it was possible to determine the interval between the first episode of KD and HLH presentation and diagnosis: in 2 cases this interval was 2 days and in the third case it was 4 days, similar to the intervals in our study. While the clinical presentations of HLH [19, 20] and HLH-KD are similar, our laboratory findings highlight several crucial differences. Compared with patients with other forms of HLH (excluding classical secondary HLH), our patients had lower incidence of anemia (25% vs. 79%), thrombocytopenia (41% vs. 97%), neutropenia (8% vs. 41%), hypertriglyceridemia (8% vs. 100%), and hypofibrinogenemia (33% vs. 70%). While both classic HLH and HLH-KD are associated with hyperferritinemia, there was more evidence of tissue hemophagocytosis (in the bone marrow, spleen, and the lymph nodes) in our study than in classic HLH (91% vs. 64%) [15].
A previous nationwide survey [21] by the Korean Society of Hematology carried out to assess the outcomes and the prognostic factors in Korean children with HLH enrolled 69 children from 21 institutions and reported an overall survival rate of 71.5%. In 2 separate surveys [21], the survival rate of patients treated with the HLH-1994 regimen was 69.1% (27/39) and 83.3% (25/30) with HLH-2004. A univariate analysis revealed that high triglyceride (>500 mg/dL), high ferritin (>6,000 µg/L), and high ALT (>500 U/L) levels were associated with poor outcomes [21]. The surveys also investigated the predominant causative gene mutations associated with familial HLH [22]. Although we were unable to evaluate the prognostic factors, the overall survival rate of the HLH-KD patients in the present study who were treated with chemotherapy alone without stem cell transplantation was 70.3%, which corroborates the findings of the nationwide survey.
In conclusion, a diagnosis of HLH-KD should be considered when manifestations similar to recurrent KD develop within 1 month of the first episode of KD. This is the first nationwide study to document the clinical characteristics of HLH-KD; its findings could help physicians differentiate between HLH and the recurrent form of KD.
 XML Download
XML Download