

 PDF
PDF ePub
ePub Citation
Citation Print
Print


INTRODUCTION
Intrinsic red blood cell (RBC) defects can result from a variety of different inherited genetic mutations as well as from acquired nutritional deficiencies that affect the structural proteins of RBCs, globin chain synthesis, and normal expression of intracellular enzymes. These disorders are typically associated with premature RBC destruction and anemia.
Hereditary spherocytosis is the most common inherited hemolytic anemia (HHA). Hereditary spherocytosis is characterized by defects in the cytoskeletal protein, spectrin, or proteins that contribute to the attachment of spectrin to the plasma membrane. This disorder results in osmotically fragile spheroidal cells that are selectively trapped in the spleen [1].
In the past, hemoglobinopathies were rarely reported in Korea. However, the number of patients with HHA is thought to have increased in recent years because of the influx of people from other South-East Asian populations and because of the increased number of interracial marriages in Korea. In addition, novel molecular techniques have simplified the methods for detecting globin gene mutations.
RBC enzymopathies can be distinguished from hereditary spherocytosis using normal osmotic fragility tests with fresh erythrocytes or by family genetic analysis. Patients with RBC enzymopathy do not have spherocytes on their peripheral blood smears and show a partial therapeutic response to splenectomy. Currently, there is no simple and convenient therapeutic laboratory screening test for glycolytic enzymopathy, and the first step in the diagnosis of this disorder is through the elimination of easily detected causes of hemolysis, such as spherocytosis and hemoglobinopathies [2].
A standard diagnostic approach for the accurate diagnosis of HHA is warranted since 2 nationwide epidemiologic and clinical studies in Korea, conducted from 1981 to 1990 [3] and from 1997 to 2006 [4], respectively, showed an increase in the prevalence of hemoglobinopathies and RBC enzymopathies. To address this, a working party for HHA was established by the Korean Hematology Association. This working party introduced a standard operating procedure for the diagnosis of HHA. Here, we report the epidemiologic and clinical data of patients who were diagnosed with HHA since the introduction of the standard operating procedure recommended by the Korean HHA Working Party of the Korean Hematology Association.
MATERIALS AND METHODS
The medical records of patients diagnosed with HHA using the standard operating procedure recommended by the Korean HHA Working Party of the Korean Hematology Association were analyzed. A survey questionnaire, which assessed the information of the medical records, was designed by the members of the Korean Hematology Association. This survey included only patients 15 years of age and younger. This study was approved by the Research Ethics Committee of Yeungnam University Hospital (IRB No. PCR-07-44).
Evidences of splenomegaly, gallstones, and cholangitis, as well as other relevant physical findings, and a family history of anemia were determined using the questionnaire. Initial laboratory test results included full blood counts with RBC indices, blood smear analyses, reticulocyte counts, iron studies, bilirubin concentration, Coombs test, haptoglobin, lactate dehydrogenase, and hemoglobin electrophoresis. If RBC membranopathy was suspected, the next diagnostic step was an osmotic fragility test at room temperature or after incubation of the blood sample, followed by an autohemolysis test. RBC membrane protein analysis using sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) was further used as a diagnostic method in patients for whom diagnosis was not definite or if the treating physician chose to include this analysis in the diagnostic work-up. Tests to establish a diagnosis of thalassemia included Heinz body and hemoglobin (Hb) H and Hb F analyses, as well as glycerol lysis, heat instability, and isopropanol precipitation tests. Direct sequencing and deletion analysis (multiple ligation-dependent probe amplification, MLPA) of the globin gene were then performed to ascertain the presence of a globin gene mutation [5]. A quantitative assay of the activity of RBC enzyme was carried out at a single central laboratory.
Statistical analysis
Data analyses were performed using SPSS version 13. Differences between subgroups were assessed using the 2-sided log-rank test. Differences between means in the different subgroups were analyzed using 1-way analysis of variance (ANOVA). The Kruskal-Wallis non-parametric test was used to compare values of non-Gaussian variables. The chi-square test was used to analyze non-continuous variables between groups.
RESULTS
Patient demographics
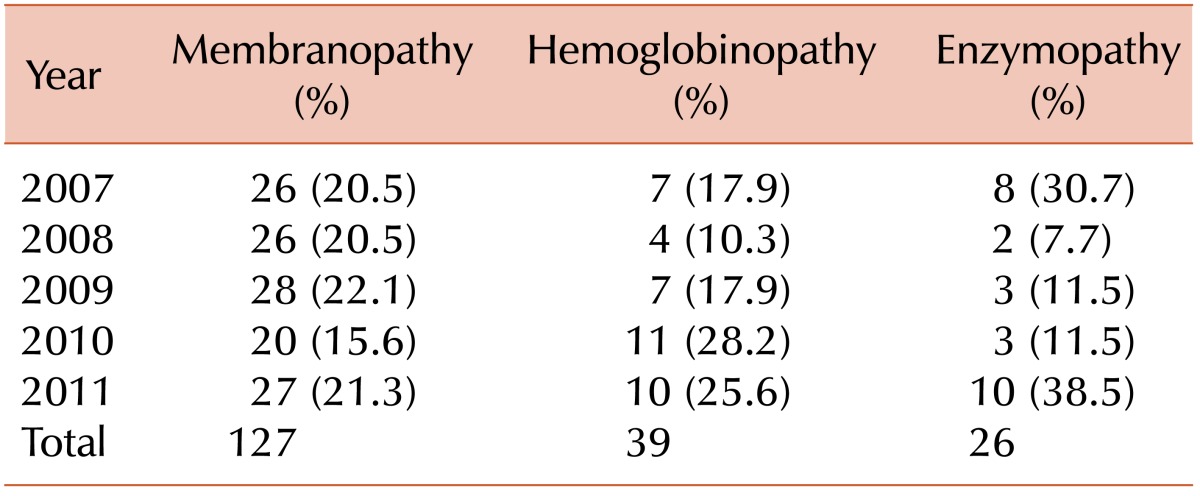
From January 2007 to December 2011, 195 patients (121 boys and 74 girls) diagnosed with HHA were registered in this survey. The median age at diagnosis was 32 months (range, 0-187 months). The annual incidences of the different disorders associated with HHA are shown in Table 1. Serial increases in the number of patients diagnosed with RBC membranopathy, hemoglobinopathy, and RBC enzymopathy were observed; the only exception was the number of patients diagnosed with RBC membranopathies in 2010, which was lower than that of previous years.
Etiology
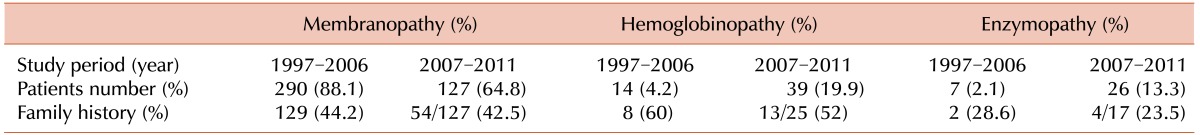
The most common cause of HHA was RBC membranopathies, which were diagnosed in 127 patients (64.8%). A diagnosis of hemoglobinopathy and RBC enzymopathy was confirmed in 39 (19.9%) and 26 (13.3%) patients, respectively. In 3 patients (1.5%), cause of HHA could not be found. The proportion of patients diagnosed with hemoglobinopathy or RBC enzymopathy was considerably higher as compared to that from the previous survey report, which assessed the prevalence of HHA from 1997 to 2006 (Table 2).
Hemoglobinopathies were the second most common cause of HHA. A globin gene mutation was confirmed in 26 patients, of which α- and β-thalassemias were present in 5 and 20 patients, respectively, and 1 patient had unstable hemoglobin disease. In the remaining 13 patients, a diagnosis of hemoglobinopathy was made on the basis of clinical and laboratory findings only. Details regarding globin gene mutations are listed in Table 3.
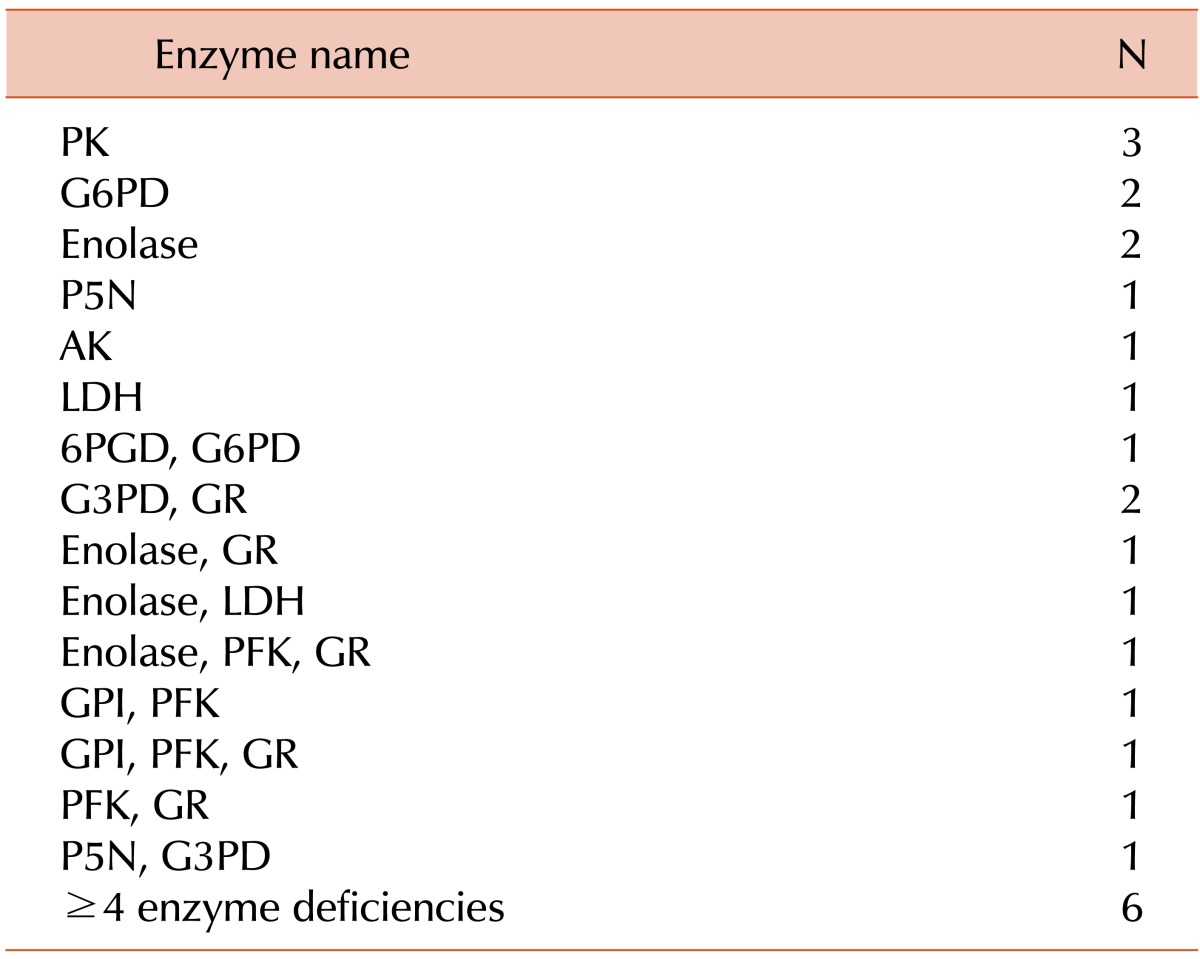
In addition, 26 patients were diagnosed with RBC enzymopathy. Isolated enzyme deficiencies were found in 10 patients; these included pyruvate kinase deficiency (3 patients), glucose 6-phosphate dehydrogenase deficiency (2 patients), enolase deficiency (2 patients), pyrimidine 5'-nucleotidase deficiency (1 patients), adenylate kinase deficiency (1 patient), and lactate dehydrogenase deficiency (1 patient). The remaining 16 patients had more than 2 enzymes deficiencies. Deficient RBC enzymes in each patient with RBC enzymopathy are summarized in Table 4.
Family history
A family history of anemia was assessed in 169 of the 195 patients. A family history indicative of HHA was established in 71 patients (42%). RBC membranopathy, hemoglobinopathy, and RBC enzymopathy were confirmed in 54 of 127 (42.5%), 13 of 25 (52%), and 4 of 17 (23.5%) patients, respectively (Table 2).
Symptoms and signs at diagnosis
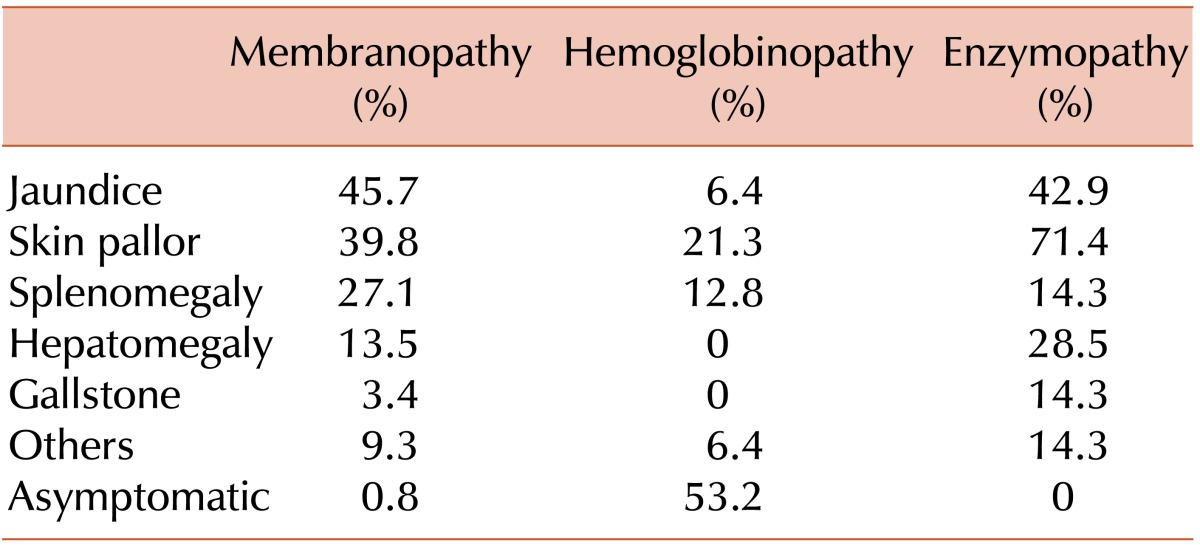
The following were considered as symptoms and signs of RBC membranopathy: anemia, jaundice, splenomegaly, hepatomegaly, abdominal distension, and gallstones. The frequency of these symptoms was similar to that reported in the 1997-2006 survey [4]. The symptoms and signs of patients with hemoglobinopathies were asymptomatic, anemia, and jaundice. Anemia and jaundice were the most common symptoms of patients with RBC enzymopathies, occurring in 71.4% and 42.9% of patients, respectively (Table 5).
Laboratory findings
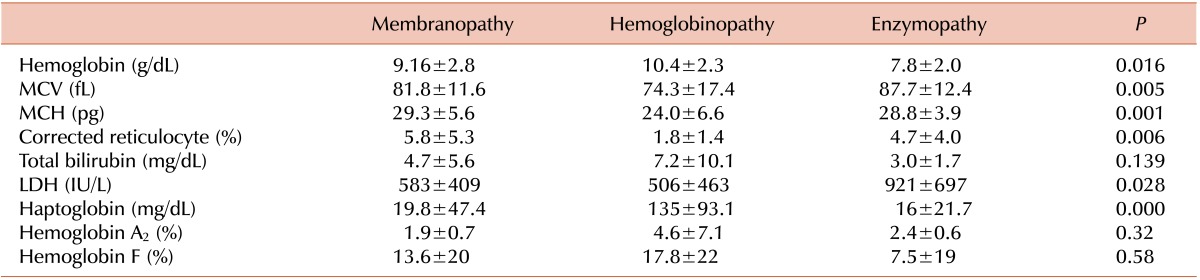
Laboratory findings for each disease subtype at the time of diagnosis are shown in Table 6. Briefly, hemoglobin concentration was lowest in patients with RBC enzymopathies (7.8 g/dL, P=0.016). Mean corpuscular volume and mean corpuscular hemoglobin concentration were lowest in patients with hemoglobinopathies (MCV 74.3 fL; P=0.005 and MCH 24.0 pg, P=0.001, respectively).
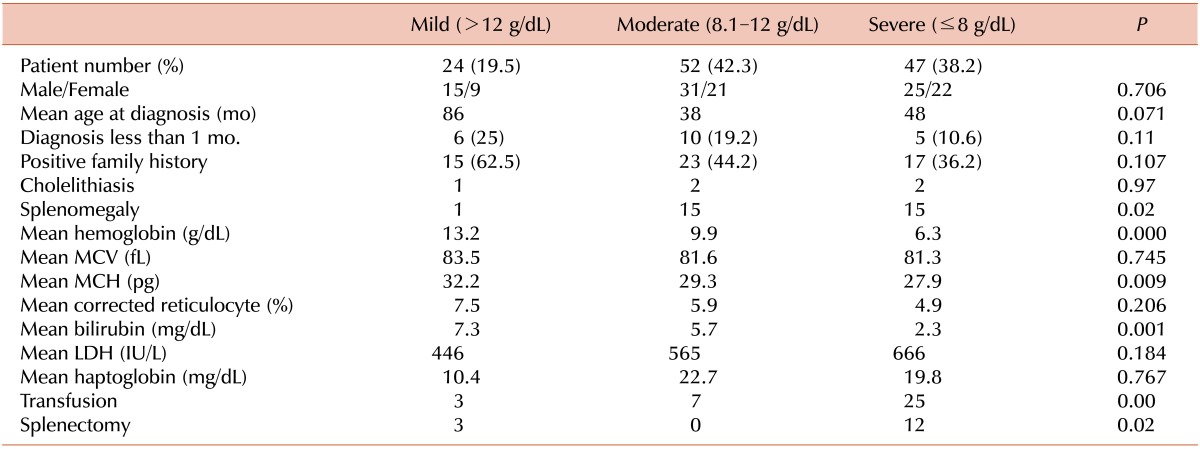
We classified 123 of the 127 patients with RBC membranopathies into subgroups of disease severity (Table 7). The 4 remaining patients were excluded because of missing laboratory findings. Hemoglobin concentration of more than 12 g/dL was defined as "mild"; 8.1-12 g/dL as "moderate"; and ≤8 g/dL as "severe." Thirty-eight percent of patients were classified as having a severe RBC membranopathy. There were no differences in age, gender ratio, family history, and frequency of cholelithiasis between subgroups. The frequency of splenomegaly was significantly higher among patients in the "moderate" and "severe" subgroups than in the "mild" subgroup (P=0.02). The total bilirubin concentration was highest in the "mild" subgroup (P=0.001). This may be related to the fact that more patients were diagnosed in the early neonatal period (25%) in this subgroup. The frequency of RBC transfusion (P<0.001) and splenectomy (P=0.02) were significantly different between groups.
Osmotic fragility tests were performed in 60 patients, and flow cytometric analyses were performed using blood samples of 8 patients from 1 institution. We did not have osmotic fragility data for the remaining 59 patients. The average start and end points of the % sodium chloride were 0.56±0.13 and 0.33±0.12, respectively.
RBC membrane protein analyses were performed by SDS-PAGE on the blood samples of 26 patients. Ten of these patients had abnormal membrane proteins, and all patients had a low ratio of spectrin to band 3 protein.
Most of the patients with β-thalassemia had mild anemia (Hb level ≥8 g/dL), except 2 patients who had severe anemia (Hb level <8 g/dL). Five patients were diagnosed with α-thalassemia, 3 of which were born to women from Southeast Asia. One patient with an ATR-16 deletion (--/αα) also had mental retardation and epilepsy, and was therefore considered as having contiguous gene syndrome (α-thalassemia with mental retardation).
The laboratory findings of hemoglobinopathies are shown in Table 6.
Treatment
Folate therapy was given to 71 of the 123 (57.7%) patients with RBC membranopathy. Splenectomy was performed in 15 of 123 (12.2%) patients, and the median age at the time of splenectomy was 7.8 years (range, 5-17 years). For the other disorders, splenectomy was not required. Transfusion was required in 28.4% (35/123) of the patients with RBC membranopathies, 10% (2/20) of the patients with hemoglobinopathies, and 60% (9/15) of the patients with RBC enzymopathies.
DISCUSSION
This study surveyed the prevalence of HHA in Korea during the period between 2007 and 2011. By comparing our findings with the data of a previous study period (1997-2006), we noticed 2 clear differences. Firstly, there was a dramatic increase in the frequency of hemoglobinopathy diagnoses (4.2% for 1997-2006 vs. 19.9% for 2007-2011) and RBC enzymopathy (2.1% for 1997-2006 vs. 13.3% for 2007-2011). Secondly, there was a relative decrease in the frequency of undiagnosed cases (6.6% for 1997-2006 vs. 1.5% for 2007-2011). In addition, the number of institutes involved in the survey decreased from 30 to 25, and enrolled patients were limited to those younger than 15 years of age in the current study. The previous study spanned 10 years and enrolled 333 patients, while the period of the present study was 5 years with 195 enrolled patients. Therefore, relatively more patients per year were analyzed in this study.
In the present study, a diagnosis of RBC membranopathy (64.8%) was the leading cause of HHA, and a large percentage of patients (38%) had severe RBC membranopathy. Approximately 60-70% of patients were classified in the group with moderate severity, followed by mild severity (20-30%), and finally sever group (5-10%) [6]. This means that the majority of patients in our survey were categorized in the moderate to severe severity groups (80%). Therefore, we think it is likely that there are a large number of people in the population with undiagnosed HHA with mild severity, suggesting that more attention is needed to identify patients with hemolytic anemia.
In 1 institution, flow cytometry-based tests as well as osmotic fragility, direct antiglobulin, and eosin 5-maleimide (EMA) binding tests were conducted to diagnose hemolytic anemia in patients with conflicting results. The data suggests that flow cytometry may be an alternative diagnostic tool that can provide additional information in the differential diagnosis of hemolytic anemia with spherocytosis [7]. The British Committee for Standards in Hematology recommends screening tests with high predictive value, such as the cryohemolysis or the EMA binding test [8]. The results of osmotic fragility tests can be affected by elevated reticulocyte counts, and the sensitivity is low. There was a report that only 66% of non-splenectomized patients with hereditary spherocytosis had increased osmotic fragility in addition to those with immune-mediated and other hemolytic anemias [9].
While almost all patients with hereditary spherocytosis can be cured with splenectomy, the decision to conduct a splenectomy should be carefully considered, and the risks and benefits should be weighed. In our cohort of younger patients with HHA, only 12% were splenectomized, and the youngest patient who underwent a splenectomy was 5 years old. The risk of cholelithiasis increases greatly after 10 years of age, but the risk of post-splenectomy infection is very high in infancy and early childhood; thus, the timing of splenectomy should be delayed until 6-9 years of age, if possible [6]. The indications for splenectomy are "moderately severe" and "severe" hereditary spherocytosis, as well as symptomatic hemolytic anemia, growth retardation, skeletal deformity, leg ulcers, and extramedullary hematopoietic tumors. Laparoscopic splenectomy is the method of choice [10, 11]. The early complications of splenectomy, such as local infection, bleeding, and pancreatitis as well as serious long-term complications, are relatively well known [12, 13]. These include postsplenectomy infection with encapsulated bacteria, an increased risk of ischemic heart disease, cerebral stroke, pulmonary hypertension, and thrombosis in later life. Long-term monitoring for late complications of splenectomy is warranted.
Genetic analysis of the globin gene is relatively simple to perform, and many patients with hemoglobinopathies can be diagnosed using this method. Because of the increased influx of people from Southeast Asia, we predict that the incidence of hemoglobinopathies in Korea may well increase in the future, although there is no substantial evidence to support this notion. In the future, large-scale screening for hemoglobinopathies targeted at second-generation children of Southeast Asian parents may be necessary.
Three different types of RBC enzymopathies, namely, glucose 6-phosphate dehydrogenase deficiency, pyruvate kinase deficiency, and enolase deficiency, were detected in a previous study in Korea. Through the present study, additional types of RBC enzymopathies were reported, and 16 of the 26 patients had more than 2 types of RBC enzyme deficiencies. Therefore, the results of quantitative assays for enzyme activity should be interpreted with caution. Enzyme activity should be estimated by assessing only surviving RBCs, without contamination of circulating white blood cells. Importantly, assays performed on blood samples may not accurately reflect in vivo enzymatic activity. Transfusion therapy in the months prior to assay analysis may obscure the presence of an enzyme defect. Reversible binding is influenced by the regulation of glycolysis, and altered binding by mutant forms of various enzymes cannot be assessed by conventional assays [2].
One of the limitations of our survey was the lack of data from adult patients. We did not include data from adult patients in our analysis because we received a limited number of completed questionnaires from adults.
In conclusion, our survey confirmed that the number of patients diagnosed with a hemoglobinopathy or RBC enzymopathy dramatically increased in Korea in the 2007-2011 period than in the 1997-2006 period. We propose standardization of the diagnostic method and the report form for the osmotic fragility test. We also advocate the introduction of more sensitive diagnostic tests to diagnose membranopathies and enzymopathies in patients with 2 or more enzyme deficiencies. We would like to emphasize that globin gene study should be considered by physicians for patients with microcytic anemia showing no evidence of iron deficiency.

 XML Download
XML Download