INTRODUCTION
The clinical manifestation of therapy-related AML (t-AML) can generally be divided into 2 groups: those who receive alkylating agents (AK) and those who receive topoisomerase II inhibitors (TI). Patients with t-AML related to AK are characterized by a long interval between therapy and AML development and are associated with cytogenetic abnormalities involving chromosome 5 and 7. Patients with t-AML related to TI present with a shorter latency and are associated with karyotype aberrations involving chromosome 11 and 21 [
1-
5].
Overall, the prognosis of t-AML is worse than
de novo AML, with a 5-year survival rate of less than 10% [
6,
7]. In contrast to
de novo AML, in which various prognostic factors have been clearly defined [
8,
9], a comprehensive study focused on the identification of independent prognostic factors in t-AML has been lacking. Although several studies have reported that karyotype is an independent prognostic parameter in t-AML, these studies focused only on cytogenetic abnormalities and did not include molecular aberrations, cytotoxic therapy regimens, or hemograms as prognostic indicators [
10-
12]. Additionally, a recently published paper reviewed clinical and cytogenetic characteristics of 39 patients diagnosed with therapy-related myeloid neoplasms from a single center, but this study did not focus on t-AML nor the prognostic factors [
13]. In this study, we performed a comprehensive evaluation to identify the independent prognostic factors in t-AML.
Go to :

MATERIALS AND METHODS
Patient selection and treatment
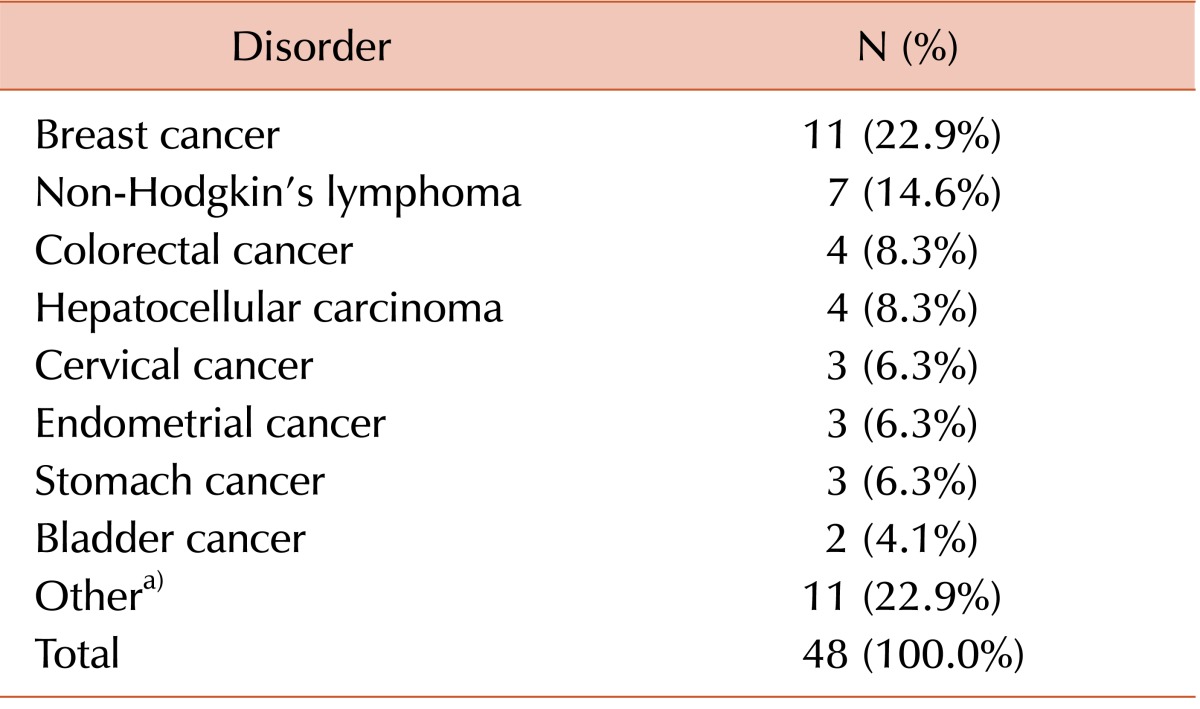
A total of 48 t-AML patients were identified in a retrospective, systematic review of electronic medical records of patients diagnosed with AML at Asan Medical Center, Seoul, Korea from January 2001 to December 2011. All patients received induction chemotherapy consisting of cytarabine and daunorubicin. The therapeutic regimen included continuous intravenous infusion of 200 mg/mL per day (100 mg/mL per day for patients aged >60 years of age) of cytarabine from day 1 to day 7, and 45 mg/mL per day of daunorubicin from day 1 to day 3. Complete remission (CR) was defined as the presence of less than 5% blasts with >20% cellularity in a bone marrow (BM) aspirate after induction chemotherapy. Relapse was defined as the presence of more than 5% leukemic blasts in bone marrow aspirates in patients with a previous CR state. Relapsed patients received the same induction chemotherapy regimen as that used at initial diagnosis. Depending on the patient's age and the availability of a suitable donor, patients received allogeneic stem cell transplantation (SCT) as close to the time of CR status confirmation as possible.
Patient classification and comparison of clinical presentation
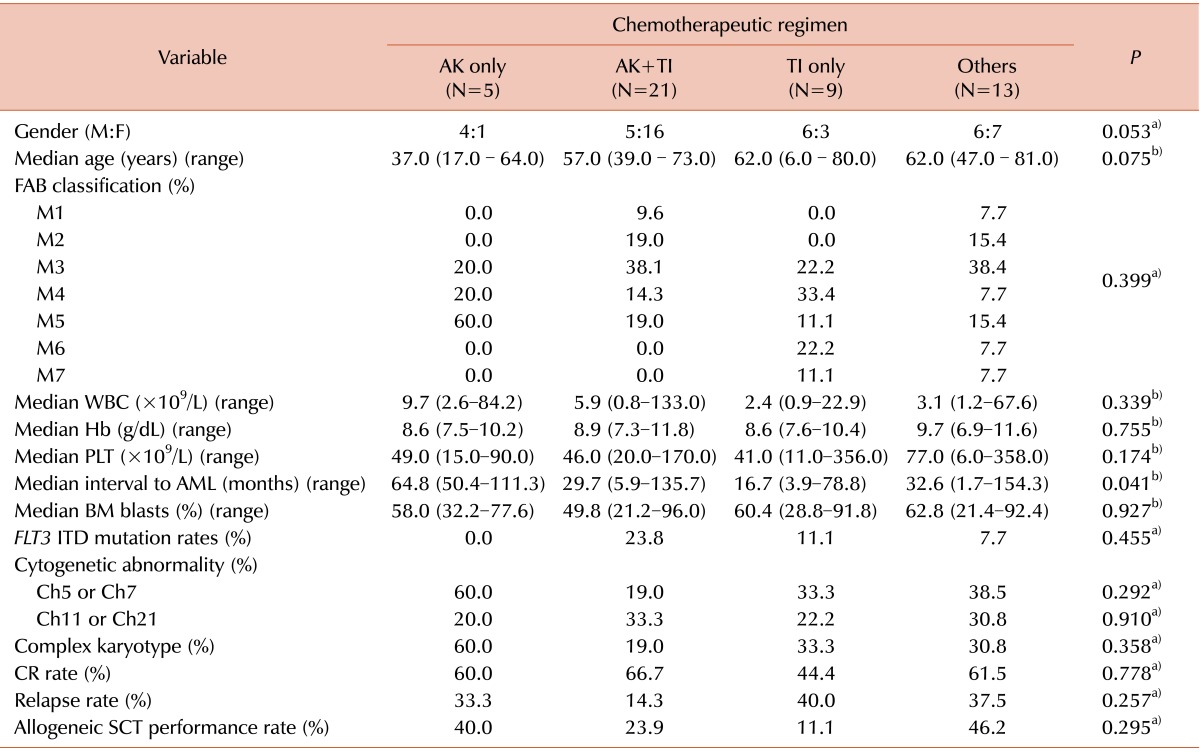
The 48 t-AML patients were categorized into 4 groups according to chemotherapeutic regimen: AK only (5 patients), AK and TI (21 patients), TI only (9 patients), and others (13 patients). Clinical data for gender, age, French-American-British (FAB) phenotype, hemogram results, FLT3 ITD mutation status, karyotype, CR rate, relapse rate, death rate, and date of allogeneic SCT were compared among the 4 patient groups. Patients who were classified as having chromosome 5/7 or 11/21 abnormalities were included in the chromosome 5/7 or 11/21 abnormality group, regardless of whether the patient had a complex karyotype or not. Similarly, patients were categorized into 2 groups with respect to the presence of a complex karyotype, irrespective of which, if any, chromosomal abnormality was present.
From these data, we calculated the prognostic markers, overall survival (OS), and disease-free survival (DFS). OS was defined as the time from diagnosis to death or, alternatively, last follow-up. DFS was defined as the time from CR to relapse for patients who experienced a relapse, death for non-relapsed patients who did not survive, or the time of the last follow-up for surviving patients who did not experience a relapse. If the patients received allogeneic SCT, the initial CR date for the calculation of DFS was replaced by the SCT date.
Comparison of prognosis
The impact of clinical variables on OS and DFS was evaluated separately. Variables included in the analysis were chemotherapy regimens, BM blast percentage, FLT3 ITD mutation status, presence of chromosome 5/7 or 11/21 abnormalities, presence of a complex karyotype, presence of a FAB M3 phenotype, and performance status of allogeneic SCT. The BM blast cell percentage comparison of prognosis was analyzed using the median value of BM blasts (61.1%) as a cutoff for the differentiation of 2 groups. Multivariate analysis was performed to confirm variables that had a significant independent prognostic impact during univariate analysis. Age, BM blasts, performance status of allogeneic SCT, FLT3 ITD mutation status, presence of chromosome 5/7 abnormalities, presence of a complex karyotype, and FAB M3 phenotype were included as covariables in the multivariate analysis.
Statistical analysis
Pearson chi-square or Fisher's exact tests were used to compare categorical variables with respect to the 4 chemotherapy groups. The Kruskal-Wallis test was used to compare continuous variables with respect to the 4 chemotherapy groups. Estimates of survival (OS and DFS) were made using the Kaplan-Meier method and were compared using a log-rank test. Multivariate analyses of OS and DFS were performed using Cox's proportional hazards model. For all analyses, tests were two-tailed and P values ≤ 0.05 were considered statistically significant. All calculations were performed using SPSS 13.0.1 for Windows (SPSS Inc., Chicago, IL, USA).
Go to :
DISCUSSION
In
de novo AML development, specific cytogenetic abnormalities and molecular aberrations such as
FLT3 ITD,
NPM1, and
CEBPA mutations have been firmly identified as prognostic indicators. In addition,
DNMT3A,
IDH1, and
IDH2 mutations are currently believed to have a potential prognostic significance [
8,
9]. In contrast, studies focused on the identification of prognostic markers in t-AML have been limited and, except for karyotype, very little information is available [
10-
12]. In our study, we aimed to identify independent prognostic factors in t-AML while taking into consideration various clinical parameters as well as molecular and cytogenetic aberrations.
In our study, the median latency from the time of diagnosis of a primary neoplastic disorder to the development of t-AML was 36.3 months, which is shorter than that reported in previous studies (median, 47.50 months) [
12,
13]. Moreover, our data showed that the median latency of t-AML as 64.8 and 16.7 months in patients receiving AK only and TI only, respectively. Given that the use of AK and TI correlates with chromosomal 5 or 7 and 11 or 21 abnormalities, our results support the previous concept that the latency of t-AML differs with respect to the chemotherapy regimen used [
14]. In our patient cohort, chromosome 5 or 7 abnormalities were detected in 15/48 (31.3%) t-AML patients; among them, 14 (93.3%) showed either a whole or partial deletion of chromosome 5 or 7. Chromosome 11 or 21 abnormalities were present in 14/48 (29.2%) of t-AML patients, and 10 (71.4%) of these patients showed a balanced translocation involving 11q23. These results also correlate with previous studies [
13,
14].
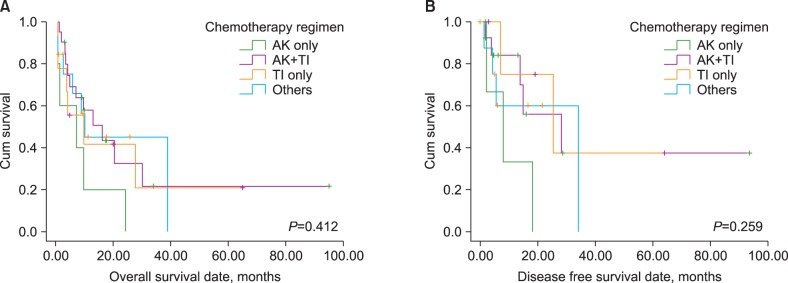
Previous studies that analyzed prognostic factors in t-AML concluded that karyotype was the most important prognostic parameter, and both favorable and unfavorable karyotypes have been identified, which have the same prognostic impact for t-AML as that for their
de novo counterparts [
11-
14]. Our study demonstrated that there are no prognostic differences with respect to chemotherapy regimens in patients with t-AML. BM blast cell percentages and
FLT3 ITD mutation status, which were known to have significant prognostic impact in patients with
de novo AML, also did not possess any prognostic impact in t-AML patients. Despite the apparently poor prognostic impact of increasing age and favorable performance of allogeneic SCT in
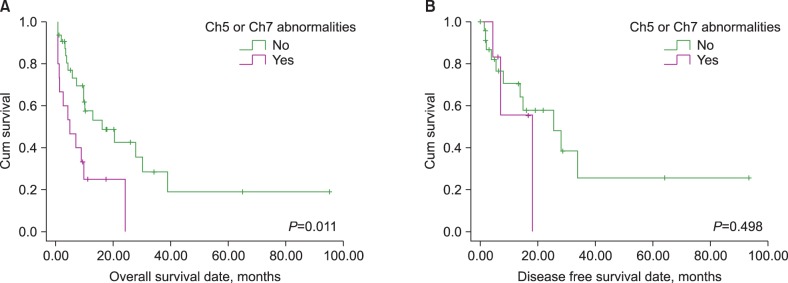
de novo AML, the present study indicated that age did not have any prognostic impact on either OS or DFS. Additionally, the performance of allogeneic SCT was only found to be a favorable prognostic indicator for OS. Although the presence of chromosome 5 or 7 abnormalities, a complex karyotype, and FAB M3 phenotype had significant prognostic value in the univariate analysis (the former two are poor and the latter one is a good prognostic indicator), which is similar to
de novo AML, multivariate analysis did not indicate that chromosome 5 or 7 abnormalities correlated with a poor prognosis. These results suggest that t-AML might have unique prognostic features compared to
de novo AML, and the establishment of a specific prognostic model for t-AML may be required.
Although data from an international study on prognostic factors in t-AML is available, an analysis focused on t-AML in the Korean population has been limited to 2 studies that reviewed demographic and clinical findings in t-AML/MDS (myelodysplastic syndrome) patients [
13,
15]. A study that focused on 39 patients with t-AML/MDS reported that breast cancer was the most common primary solid tumor (23.1%). The same study also demonstrated a shorter latency interval in patients with balanced rearrangements than in patients with the loss of chromosome 5 or 7 [
13]. These findings are consistent with the results of the present study; however, the previous study reported that balanced translocations were frequently detected in patients who had undergone therapy to treat a solid tumor and that most patients with balanced translocations developed t-AML rather than t-MDS [
13]. This trend could not be confirmed in the present study, since our study population did not include t-MDS patients. Another study focused on 12 patients who developed therapy-related acute leukemia after treatment for breast cancer and reported that 67% of patients had balanced translocations involving 11q23 and that patients with 11q23 translocation showed markedly poor event free survival than those without 11q23 translocation [
15]. Contrary to these results, the presence of chromosome 11 or 21 abnormalities did not possess a significant prognostic impact on survival in our study, although the latency of AML transformation was significantly shorter than that without chromosome 11 or 21 abnormalities. The prognostic impact of the presence of chromosome 11 or 21 abnormalities, including 11q23 translocations should be confirmed with a larger study population.
Considering the FAB AML M3 phenotype, our patient cohort included 8 patients who manifested acute promyelocytic leukemia (APL). All of these patients possessed a
PML/RARa fusion transcript, which was demonstrated by reverse transcriptase PCR. Notably, we found that 4 of the 8 APL patients (50.0%) had an additional, single cytogenetic abnormality (inv(6) for 1 patient, del(7) for 1 patient, and +8 for 2 patients). This frequency is higher than that found in previous studies, which report an incidence of 25.41% [
16-
20]. Given that the frequency of an additional cytogenetic abnormality in our non-APL patients was 26/40 (65.0%), which was not significantly different (
P=0.692), these results suggest that the good prognosis of an APL manifestation of t-AML might be explained by its unique characteristics, and not due to a low frequency of additional cytogenetic abnormalities. Our APL patients had unique clinical manifestations such as longer latency of t-AML (mean, 59.2 months) and a higher CR rate (87.5%) than that reported in previous studies (latency of 2.3 years and CR rates of 60.80%) [
16-
19]. These results may be additional evidence for the presence of unique clinical features such as very good prognosis for t-AML patients who manifest with APL. Given that the prognosis of t-AML patients who manifest with APL proved to be better than the other subtypes in the present study, the prognostic impact of the presence of t(8;21) or inv(16) at the time of t-AML diagnosis (a good prognostic factor in
de novo AML) is an interesting question that should be further investigated. However, this issue could not be addressed in the present study, because it included only 1 patient with t(8;21) and no patients with inv(16) at the time of t-AML diagnosis. A larger study population would be required evaluate this issue.
Our study had some limitations. First, several molecular prognostic markers such as mutations in NPM1, CEBPA, DNMT3A, IDH1, and IDH2 were not evaluated. Since the prognostic value of these markers has been established in patients with normal karyotype de novo AML, this limitation may have negatively influenced the results of this study. Second, the number of patients included in each group was relatively small, which may have affected the statistical power of our study. A comprehensive study with an adequate number of patients is required to confirm our hypothesis.
In conclusion, patients with a t-AML M3 phenotype were identified as having a better prognosis than other subtypes, suggesting that the AML M3 phenotype possesses unique clinical features. Among the possible prognostic factors, karyotype was the strongest prognostic indicator, predicting a poor prognosis for t-AML patients with a complex karyotype.
Go to :


 PDF
PDF ePub
ePub Citation
Citation Print
Print


 XML Download
XML Download