

 PDF
PDF ePub
ePub Citation
Citation Print
Print


INTRODUCTION
Graft-versus-host disease (GVHD) is one of the main causes of death in patients undergoing allogeneic hematopoietic stem cell transplantation (HSCT) in hematologic malignancies, including acute myeloid leukemia (AML), acute lymphocytic leukemia (ALL), and myelodysplastic syndromes (MDS) [1, 2]. GVHD can be divided into acute GVHD (aGVHD) and chronic GVHD (cGVHD). aGVHD is caused by donor T cell-mediated recognition in response to recipient nonhematopoietic antigen presenting cells, especially dendritic cells. In consequence, in the immune reaction, alloantigens are presented to alloreactive T cells with huge amounts of cytokines [3, 4]. Unlike aGVHD, cGVHD is generated by thymic damage, production of aberrant B cells, defective function of T cells, along with cytokine dysregulation [5]. Numerous approaches have been applied as part of transplantation protocols in order to inhibit the occurrence of GVHD, such as T cell depleted donor grafts, pharmacological agents, infusion of regulatory T cells, and gene expression profiling of T cell subsets from donors [6-10]. However, the incidence of aGVHD remains high, at over 60%, while 50-70% of recipients develop cGVHD [2, 11].
Recently, Nishimori et al. [8] demonstrated that immune cell-related cytokines, which aid differentiation into effector T cells (e.g. Th1, Th2, and Th17), may also function as key modulators in GVHD [8, 12, 13], and their activities can be attenuated by suppressor of cytokine signaling (SOCS) proteins [14]. SOCS proteins are inhibitors of cytokine signaling pathways and are key physiological regulators of both innate and adaptive immunity. The cytokine inducible SH2-containing (CIS) SOCS family contains 8 members (CIS and SOCS1-SOCS7), each of which has a central SH2 domain, an N-terminal domain of variable length and sequence, and a 40-amino-acid C-terminal module called the SOCS box [15]. In particular, the role of SOCS1 and SOCS3 in Toll-like receptor immune responses has been extensively investigated [16, 17]. Furthermore, previous studies have shown the potential for enhancing T-cells in tumors by high-level transcription of SOCS genes that result in the inhibition of SOCS proteins and cytokines, including interferon gamma (IFN-γ) [18, 19]. Knockout experiments with SOCS1-deficient mice revealed that SOCS are linked to immune-related cytokines, such as IFN-γ and interleukin 6, and to defects in T cell homeostasis [20]. Moreover, SOCS are also key regulators of aGVHD pathology via a cytokine storm and act to enhance Th1 cell activation [21]. Of particular note, SOCS genes have well-documented therapeutic effects and are therefore promising candidates for the treatment of hematologic malignancies, such as leukemia and solid-organ transplantation [22-24].
Despite increasing evidence for the importance of SOCS in governing immune mechanisms to control GVHD, whether SOCS are coordinately expressed in recipients after allogeneic HSCT remains unknown. In this study, we investigated the expressions of SOCS1 and SOCS3 in adult recipients with aGVHD and cGVHD who received allogeneic HSCT, and examined the feasibility of SOCS as promising therapeutic targets and prognostic predictors in GVHD.
MATERIALS AND METHODS
Human blood sampling and preparation
All experiments were performed with authorization from the Institutional Review Board for Human Research at the Catholic University of Korea. All blood samples were collected from post-HSCT recipients, who were initially diagnosed with one of the hematologic diseases designated by the World Health Organization. In addition, peripheral blood was donated from a set of healthy transplant donors (N=55). Heparinized blood samples were obtained from all transplant recipients within 1 week of GVHD development, and on the day of transplantation from all donors. Mononuclear cells were isolated by overlaying the blood samples on a Ficoll-Hypaque gradient (density, 1.077; Lymphoprep; Gibco-BRL, Carlsbad, CA, USA), followed by centrifugation at 400 ×g for 30 min. The buffy coats were harvested and washed twice with phosphate-buffered saline (pH 7.4).
Clinical characteristics
Clinical characteristics of the recipients and donors enrolled in this study are detailed in Table 1. A total of 71 recipients with AML (N=40), ALL (N=12), MDS (N=10), chronic myelogenous leukemia (CML; N=2), severe aplastic anemia (SAA; N=5), or others (N=2), who received allogeneic HSCT from human leukocyte antigen-identical siblings or unrelated donors between 2009 and 2011, were included in the present study.
Clinical record and GVHD grading
Diagnoses of aGVHD and cGVHDs were determined as described previously, based on consensus criteria [25, 26]. The classification of aGVHD was determined by its severity as no (none GVHD and grade I), grade II, and grade III-IV. Based on clinical impressions of its overall severity, cGVHD was classified from mild-moderate to severe. Recipients without GVHD after HSCT were classified into the none-GVHD group. Methylprednisolone was administered at 2.4 mg/kg/day for 4.7 days with a gradual taper to treat aGVHD graded II or more. Skin, rectal, stomach, or duodenal biopsies were performed in order to confirm the GVHD diagnoses [27]. The treatment of cGVHD was also variable; in accordance with National Institute of Health recommendations, the mild type was treated with topical immunosuppressants, whereas both moderate and severe types were treated with a calcineurin inhibitor and systemic steroids [28].
Real-time quantitative reverse transcription PCR (qRT- PCR) analysis
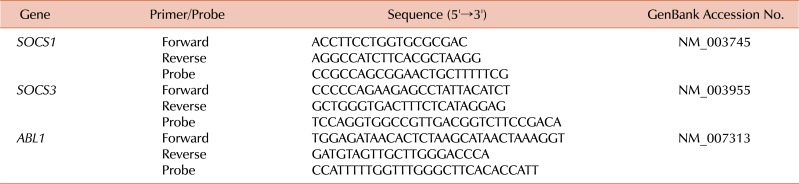
Since no data regarding the levels of SOCS genes of the recipients were available, we performed qRT-PCR of the blood samples from all recipients and donors in this study in order to investigate whether SOCS1 and SOCS3 expression levels were associated with any post-HSCT complications, such as aGVHD and cGVHD. Total RNA was extracted from mononuclear cells using the TRIzol reagent (Invitrogen, Carlsbad, CA, USA). The RNA samples were treated with RNase-free recombinant DNase I (Roche, Mannheim, Germany) and subjected to reverse transcription using the Transcriptor First-Strand cDNA Synthesis kit (Roche). cDNA synthesis was achieved by incubating at 25℃ for 10 min and at 42℃ for 60 min, after which the reaction was inactivated by heating at 99℃ for 5 min. The qRT-PCR reactions and fluorescence measurements were performed using a LightCycler 480 Real-Time PCR system (Roche). The probe was labeled at its 5' end with 6-carboxy-fluorescein reporter dye and at its 3' end with 6-carboxy-tetramethylrhodamine, as a quencher. The qRT-PCR primers and probes for SOCS1, SOCS3, and ABL1 (control gene used for normalization), are listed in Table 2. Quantitative amplification was performed using the following parameters: denaturation at 95℃ for 10 min, followed by 50 cycles of denaturation at 95℃ for 10 s, and annealing and elongation at 60℃ for 30 s, with a final cooling step at 40℃ for 30 s. All the sample analyses were performed in triplicate.
Statistical analysis
All results are presented as mean±standard error of the mean (SEM) values. Statistical analyses of more than 2 groups were performed using one-way analysis of variance (ANOVA). The difference in the frequency of each gene between donors and recipients was analyzed using a two-tailed Wilcoxon matched-pairs signed rank t-test. Values of P<0.05 were deemed to indicate statistical significance. All calculations were performed using the GraphPad Prism 5 software (version 5.0.3; GraphPad Software Inc., San Diego, CA, USA).
RESULTS
Expression of SOCS1 and SOCS3
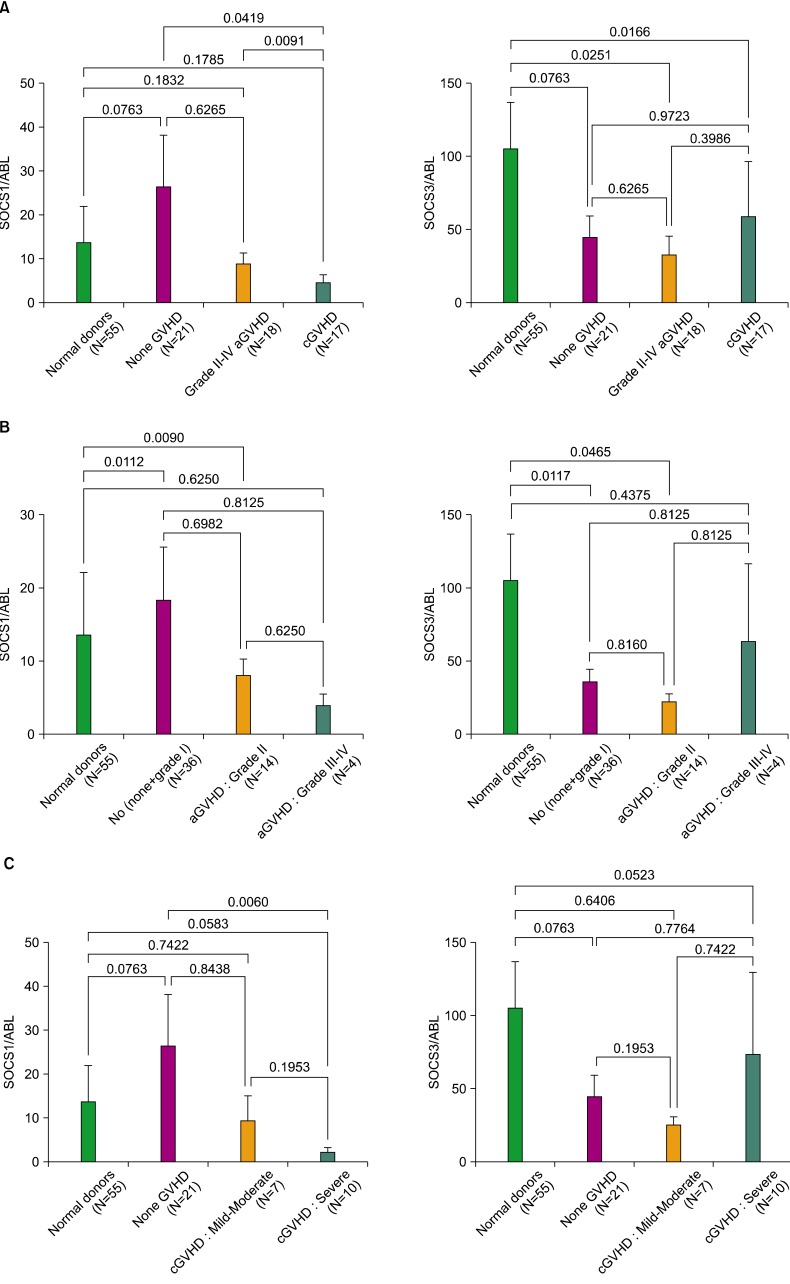
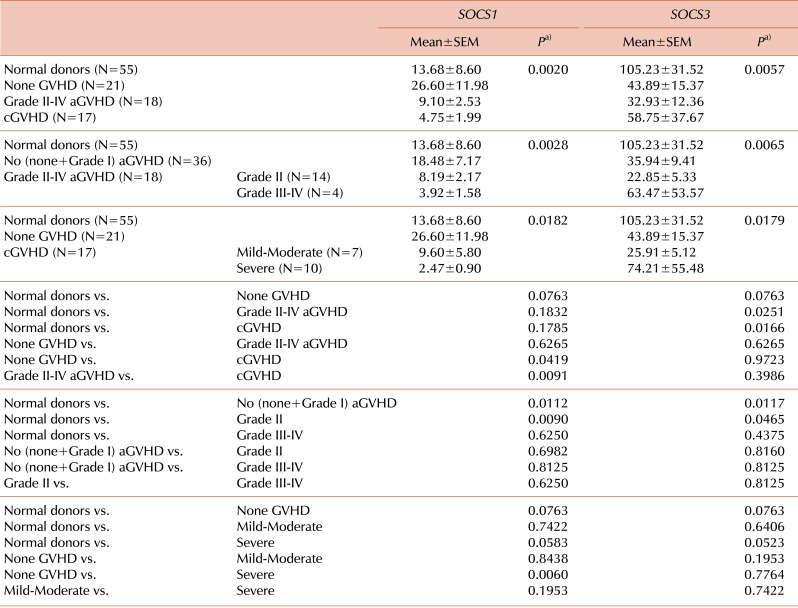
Of the 71 HSCT recipients with successful engraftment evaluated, 18 (25.4%) developed grade II to IV aGVHD: 14 (77.8%) with grade II and 4 (22.2%) with grade III-IV. cGVHD developed in 17 (23.9%) of the recipients evaluated. Our data showed that SOCS1 expression marginally increased in the none-GVHD group compared to normal donors (P=0.0763). Interestingly, SOCS3 expression decreased in the none-GVHD group (P=0.0763). Results of the one-way ANOVA showed significant effects of the SOCS1 and SOCS3 genes for the normal donor, none-GVHD, grade II-IV aGVHD, and cGVHD groups (P=0.0020 and P=0.0057, respectively). The expression level of SOCS1 decreased in the cGVHD, unlike that in the none-GVHD group (P=0.0419), and was significantly lower in cGVHD than in aGVHD recipients (P=0.0091) (Fig. 1A, left panel). Meanwhile, a statistical difference of SOCS3 expression was detected in both the aGVHD and cGVHD groups (P=0.0251 and P=0.0166, respectively), when compared to normal donors (Fig. 1A, right panel).
Expression of SOCS1 and SOCS3 based on the severity of aGVHD and cGVHD
We further analyzed the expression levels of SOCS genes in terms of the severity grade of aGVHD, in which no (none+grade I), grade II, grade III-IV, and cGVHD were graded from mild-moderate to severe. The SOCS1 and SOCS3 genes showed significant effects on GVHD severity (P=0.0028 and P=0.0065, respectively). SOCS1 expression was lower in grade II aGVHD recipients and higher in no aGVHD recipients than in the normal donors (P=0.0090 and P=0.0112, respectively; Fig. 1B, left panel). Meanwhile, SOCS3 expression was significantly lower in the grade II and no aGVHD groups than in the normal donors (P=0.0465 and P=0.0117, respectively), but showed less reduction in the grade III-IV aGVHD group (Fig. 1B, right panel).
We further investigated the expression of SOCS genes in terms of severity of cGVHD. The effects of SOCS1 and SOCS3 on cGVHD severity were significant (P=0.0186 and P=0.0179, respectively). The only significant difference in SOCS1 expression was found in the comparison between the severe cGVHD and the none-GVHD groups (P=0.0060; Fig. 1C, left panel), suggesting that severe cGVHD is closely associated with SOCS1 gene regulation. As shown, SOCS1 expression was clearly lower in the mild-moderate cGVHD subgroup than in the none-GVHD group, although this difference was not significant due to the small number of samples. We noted a marginally significant difference in SOCS1 expression levels between severe cGVHD and normal donors (P=0.0583). In contrast, SOCS3 expression did not differ between any of the groups compared, except for a marginally significant difference between severe cGVHD and normal donors (P=0.0523; Fig. 1C, right panel). Overall, SOCS3 expression decreased relatively less with increasing severity of both aGVHD and cGVHD, implying the presence of a highly dynamic molecular interaction between the development of GVHD and the regulation of SOCS1/SOCS3 genes post-HSCT.
DISCUSSION
Prevention of GVHD after allogeneic HSCT has been extensively investigated in the past decade [6-9]. A successful engraftment of normal hematopoietic stem cells without rejection and/or severe GVHD should be a goal for all procedures involving allogeneic HSCT. To achieve this, various cytokines that can regulate donor-derived T cell immune activation need to be balanced prior to GVHD occurrence after HSCT [13]. In particular, immune suppressor cytokines, which are regulated by SOCS family genes, may play an important role in the various pathophysiological events that occur following allogeneic HSCT, as indicated by animal studies [21, 24]. Results of previous animal studies on the role of SOCS genes led us to question whether some of these negative regulators of cytokines, SOCS1 and SOCS3, may be involved in the development of GVHD by T- and/or B-cell activation. Understanding how SOCS genes are differently regulated across various subsets of disease severity would be essential for the development of therapeutic targets for treatment.
We found that SOCS1 and SOCS3 expression levels were differentially regulated after allogeneic HSCT. These data were all obtained independently from qRT-PCR experiments, and all genes were normalized to the ABL1 gene in this study, as shown in Table 3. The remarkably contrasting expression levels of the 2 genes in non-GVHD recipients indicate that the molecular machinery of SOCS1 and SOCS3 genes show different interactions in terms of regulatory balance and severity of development of aGVHD and cGVHD, indicating incomplete negative feedback for cytokines in specific GVHD conditions. The relatively maintained level of SOCS1 gene expression in the none-GVHD group compared to normal donors suggests that stable expression of SOCS1 may reduce the development of GVHD by inhibiting cytokine storm, as well as sustained engraftment of normal hematopoiesis under a certain degree of regulatory power. However, SOCS3 showed the completely opposite response. In contrast to SOCS1, SOCS3 expression was markedly decreased in the none-GVHD group compared to normal donors, suggesting that different molecular mechanisms underlie the activities of SOCS1 and SOCS3, and further implying incomplete negative feedback of cytokines under particular GVHD conditions. In addition, SOCS1 expression tended to decrease with increasing severity of aGVHD and cGVHD compared to no (none+grade I) or none GVHD, which suggested that cytokine storming was induced by failure of SOCS1 stimulation in the aGVHD group, leading to rapid functional dysregulation of the interaction between cytokines and the SOCS1 gene. Although the difference was not significant, expression levels of SOCS3 showed a relatively lower pattern of decrease with increasing severity of both aGVHD and cGVHD, implying that SOCS3 is essential for the regulation of GVHD. In addition to highlighting the relevance of these genes for steroid administration in aGVHD, these results were further confirmed by using the no GVHD group as a control, which combined the none-GVHD group with the grade I group.
However, the present study is somewhat limited, as clinical relevance of these results could not be established due to the small number of patients, although drug treatments commonly start from grade II of aGVHD. More defined clinical outcomes will likely emerge with the accumulation of further studies correlating SOCS genes with the occurrence of aGVHD. Among the 18 aGVHD patients analyzed in this study, we found only 1 case of acute/chronic overlap syndrome, which was intentionally excluded to facilitate interpretation of the data. Together, these findings suggest that the SOCS1 and SOCS3 genes appear to inhibit cytokine signaling by different mechanisms in aGVHD and cGVHD. Previous GVHD studies with animal models have shown associations between SOCS1 and SOCS3 and immune disorders by T cell activation in GVHD [8, 12, 13, 21]. Although some studies have shown that SOCS3-/ΔLck T cells exacerbate scleroderma GVHD in mice in a cytokine dependent manner, suggesting the importance of SOCS3 for GVHD [21], our data showed a pattern of increased SOCS3 expression in severe cases of cGVHD. This may be due to individual variation resulting from differences in the specific medical treatments applied or in the species' genetic backgrounds. This study is the first to investigate expression patterns of both SOCS1 and SOCS3 in human GVHD recipients. Due to their suppressive properties on immune cells as demonstrated in recent studies, SOCS genes are emerging as potential therapeutic targets for several diseases, including GVHD [22-24].
Among members of the SOCS family, SOCS1 and SOCS3 have been shown to contain a kinase inhibitory receptor (KIR), based on a knockout system applied to KIR-containing SOCS members, implying the importance of KIR domains in immune systems [18, 19]. Therefore, we focused on the SOCS1 and SOCS3 genes in this study. Regulation of SOCS family proteins can occur at the transcriptional level as well as at the translational and post-translational levels. Furthermore, transcription of these genes can be initiated by cytokines, suggesting the importance of gene expression in direct cytokine regulation [18, 19]. Of particular note, recent studies have suggested that SOCS molecules might interact with other SOCS members, for example SOCS2 and CIS molecules could aid in the degradation of SOCS1 and SOCS3, or translational regression from other proteins may induce the regulation of SOCS genes, implying a counter-regulated function [29, 30]. Therefore, further study of the cross-modulation of these molecules will inform therapeutic strategies for the effective prevention of GVHD or the frequently encountered cases of aggressive GVHD progression after its initial development. Further evaluation of regulatory balances of SOCS1 and SOCS3 in specific transplant cases or under conditioning regimens, such as myeloablative conditioning (MAC), or reduced-intensity conditioning of allogeneic HSCT based on anti-thymocyte globulin, are needed to reveal the direct contribution of cytokine feedback mechanisms for GVHD. In addition, the statistically significant levels of SOCS genes in GVHD found in this study (Fig. 1B and C) should be verified in future studies using higher sample sizes, similar distributions of each subgroup of GVHD, and other influential clinical parameters. Moreover, future studies should aim to determine whether or not the expression levels of SOCS genes are tightly regulated in a severity-dependent manner in GVHD with hematologic diseases.
In conclusion, we present the first report that SOCS1 and SOCS3 are differentially expressed in recipients after allogeneic HSCT, suggesting a prognostic correlation between SOCS genes and the development of GVHD. These results, along with those of future studies, will shed light on the possibility of SOCS genes as good candidates for developing new diagnostic and therapeutic tools for GVHD after allogeneic HSCT.

 XML Download
XML Download