

 PDF
PDF ePub
ePub Citation
Citation Print
Print


INTRODUCTION
Hepcidin (hepatic bactericidal protein) was initially identified as a urinary antimicrobial peptide rich in cysteine. Further studies showed that hepcidin is overexpressed in mice with iron overload and that it plays a significant role in iron homeostasis in knockout animals with iron storage disease. The complete abrogation of hepcidin entails excessive intestinal absorption of iron and increased iron release by macrophages, a condition that leads to iron overload [1-3]. Hepcidin performs its different functions via a single biochemical mechanism: hepcidin-ferroportin interaction [4]. Intestinal epithelial cells and reticuloendothelial macrophages use the same transporter, ferroportin, to transport iron in the plasma. Moreover, macrophages and enterocytes exhibit strong upregulated ferroportin expression in the erythropoietic response in an iron-restricted state.
Go to : 
HEPCIDIN REGULATION
Iron regulates hepcidin homeostasis. Increases in iron levels in the plasma and iron storage stimulate the production of hepcidin, which blocks iron absorption from the diet and its further storage [5]. Hepcidin production is suppressed in the case of iron deficiency; the feedback loop between iron and hepcidin should ensure the appropriate physiological concentration of iron in the plasma. Hepcidin production is also regulated by the erythropoietic process, whose core activity is characterized by high iron consumption. In this case, hepcidin suppression causes the stored iron to be released by hepatocytes and macrophages while the intestinal absorption of iron increases. This condition involves iron supplementation from hemoglobin synthesis activity [6]. Hepcidin is elevated during inflammation and/or infection. This can cause iron dysregulation with hypoferremia and anemia related to inflammatory disease [7]. Hypoferremia can also represent a strategic host defense to limit iron availability to microorganisms.
Go to :
MOLECULAR MECHANISM OF HEPCIDIN REGULATION
Bone morphogenetic proteins (BMPs) are a group of growth factors that activate the transduction signal by interacting with specific receptors. They use intracellular and extracellular iron detection mechanisms to alter hepcidin expression. BMPs increase hepcidin both in vivo and in vitro. BMP6 is the primary regulator of endogenous hepcidin. Hemojuvelin (HJV) is a BMP co-receptor specialized for iron regulation [8]. The hepcidin adjustment process in the iron response occurs in the liver on the membranes of hepatocytes. Once BMP6 binds the co-receptor HJV, the SMAD transcriptional system (SMAD1/5/8) is activated [9-11]. The activated SMAD complexes bind directly to BMP-responsive elements on the hepcidin promoter, thus inducing hepcidin transcription [12].
The TFR2 and HFE genes, which are altered in adults with hereditary hemochromatosis, appear to adjust to the detection of iron-transferrin concentration. TFR2, a TfR1 homolog, is mainly expressed in the liver. HFE is similar to major histocompatibility complex type I (MHC I) molecules. Regarding the iron-transferrin concentration complex, HFE appears to function as a shuttle between TfR1 and TFR2.
Go to :
HEPCIDIN AND ERYTHROPOIESIS
Increased erythropoietic activity significantly reduces hepcidin levels. A single administration of erythropoietin (EPO) over a period of 24 hours significantly reduces hepcidin levels in humans [13]. In cases of ineffective erythropoiesis, 2 proteins are produced by erythroblasts, growth differentiation factor 15 (GDF I5) and twisted gastrulation I (TWSGI), which appear to be responsible for mediating hepcidin suppression [14, 15]. EPO indirectly influences iron homeostasis. EPO production as a normal response to hypoxic stimulation is responsible for normal erythron expansion without excessive erythropoiesis. GDF15 and TWSG1 are released as a result, ultimately suppressing hepcidin synthesis as mentioned before. EPO activation is the main event that occurs in acute hypoxia; this causes the expansion of erythropoiesis, which requires adequate iron for the hemoglobinization of red cells. The production and hemoglobinization of the erythroid lineage can still occur, if hepcidin is downregulated [16, 17].
Go to :
HEPCIDIN AND INFLAMMATION
Inflammation and infection increase hepcidin synthesis. Patients with sepsis, inflammatory bowel disease, myeloma, burns, and C reactive protein (CRP) levels >10 mg/dL exhibit significantly elevated hepcidin levels [3, 5, 7, 18, 19]. Macrophages are stimulated during the inflammatory process; the stimulation depends on the severity of inflammation. Activated macrophages release a network of cytokines. Among them is interleukin-6 (IL-6) is one of the primary inducers of hepcidin expression; an increase in hepcidin levels finally results in hypoferremia (Fig. 1). Hepcidin inhibits iron release from macrophages as well as intestinal iron absorption. In inflammatory states, hepcidin production is no longer regulated by iron burden (i.e., if the iron level is low, hepcidin synthesis should be downregulated) but is rather increased through IL-6 stimulation. Serum iron was demonstrated to affect hepcidin synthesis in healthy volunteers, in whom the early presence of hepcidin in the urine was measured after an oral iron administration dose that did not affect iron storage. Serum iron is an induction signal for hepcidin production and affects serum transferrin saturation percentage. In the case of inflammation, hepcidin can also be produced by myeloid cells via the activation of TRL4, a receptor located on the membranes of neutrophils and macrophages [20].
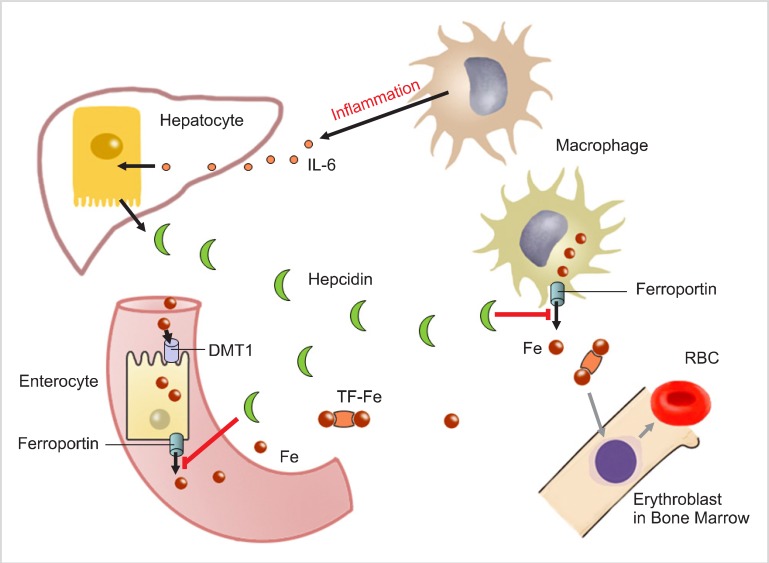
Go to :
HEPCIDIN AND ANEMIA
Understanding the physiological processes of hepcidin has made it possible to redefine the pathogenetic mechanisms of anemia.
1. Iron deficiency anemia
In pure iron deficiency anemia (IDA), serum and urinary hepcidin concentrations are significantly decreased and are even undetectable by some methods currently in use. Even in the absence of anemia, hepcidin appears to be a sensitive indicator of iron deficiency. Moreover, compared to hematocrit or hemoglobin, a decrease in hepcidin is an early marker of iron deficiency together with transferrin saturation and decreased ferritin. Since hepcidin in the urine may also be measured, samples can be collected easily from babies and children.
2. Iron-refractory iron deficiency anemia
Iron-refractory iron deficiency anemia (IRIDA) is a genetically transmitted hypochromic microcytic anemia. It is characterized by increased hepcidin production due to a gene mutation in the suppressor matriptase-2 (TMPRSS6). Extracellular BMP2, BMP4, and BMP6 bind to the co-membrane receptor m-HJV as well as BMP receptor (BMPR). This condition triggers the phosphorylation of SMAD1, SMAD5, and SMAD8 as well as the formation of heteromeric complexes with SMAD4 as the common mediator. After nuclear translocation, heteromeric SMAD complexes stimulate the transcription of the Hamp gene, which is responsible for hepcidin production. Hepcidin transcription is negatively regulated by soluble HJV (s-HJV), which acts as an antagonist of the BMP pathway, competing with m-HJV for BMP ligands. When matriptase-2 is mutated, hepcidin increases, resulting in the chronic inhibition of iron absorption and consequent anemia [21-23].
3. Anemia with iron overload
In β-thalassemia and congenital dyserythropoietic anemia, anemia is characterized by iron overload. Patients who do not receive transfusions have greatly reduced serum and urinary hepcidin levels. Increased erythropoietic activity and the lack of hepcidin adjustment due to the iron overload suppress the signal for the production of hepcidin itself. In ineffective erythropoiesis syndromes, the suppression of hepcidin production is regulated by GDF15 and TWSG I [1, 14, 15]. Hepcidin levels are much higher in chronically transfused patients than that in non-transfused patients due to iron overload and ineffective erythropoiesis [24-26]. In non-transfused thalassemic patients, iron is stored in hepatocytes rather than macrophages, similar to that in transfused thalassemic patients. The consequence of this different iron cellular distribution is that serum ferritin is much lower in non-transfused patients and does not adequately reflect liver iron storage.
4. Anemia of chronic disease/inflammation
Patients with infections, chronic inflammatory disorders, and cancers have "anemia of chronic disease/inflammation" (ACD) [27]. Hepcidin is elevated in the following inflammatory conditions: rheumatic diseases [3], inflammatory bowel disease [18], chronic infections [3], multiple myeloma [19], and critical disorders.
There is a rare form of iron-refractory hyposideremic anemia generally present in hepatic adenoma. The early removal of the adenoma resolves the hypoferremia and the consequent anemia, probably because the tumor is the cause of autonomous hepcidin production.
Obesity can also be considered a chronic inflammatory state that can cause hyposideremia [28]. In both anemia and hyposideremia, the elevated hepcidin level helps differentiate ACD from IDA. A condition of "mixed anemia" can arise in chronic inflammatory diseases involving bleeding and/or malnutrition. Under these conditions, the hyposideremia could counteract the hepcidin increase mediated by inflammation. A true iron deficiency from non-intestinal absorption by hepcidin may develop when inflammation is present for years.
5. Anemia in chronic kidney disease
In patients with anemia in chronic kidney disease (CKD), the anemia is mainly due to the lack of EPO. Kidney function (i.e., excretion) plays an important role in hepcidin clearance. Kidney dysfunction results in decreased hepcidin clearance and consequent hepcidin storage with hyposideremic anemia development [29]. As CKD progresses, hepcidin level increases regardless of the inflammatory state. Hepcidin production may be considered to mirror genetic hemochromatosis and chronic diseases (Fig. 2) [30]. Hemochromatosis is characterized by low hepcidin production, which increases intestinal absorption of iron and iron release by macrophages. This inevitably leads to progressive iron storage in tissues. In chronic diseases, high hepcidin production inhibits iron release from macrophages and intestinal absorption of iron. This consequently induces an anemic condition. The interaction between hepcidin and ferroportin determines the plasma iron transport. Hepcidin concentration is regulated by iron, erythropoietic activity, and inflammation [31].
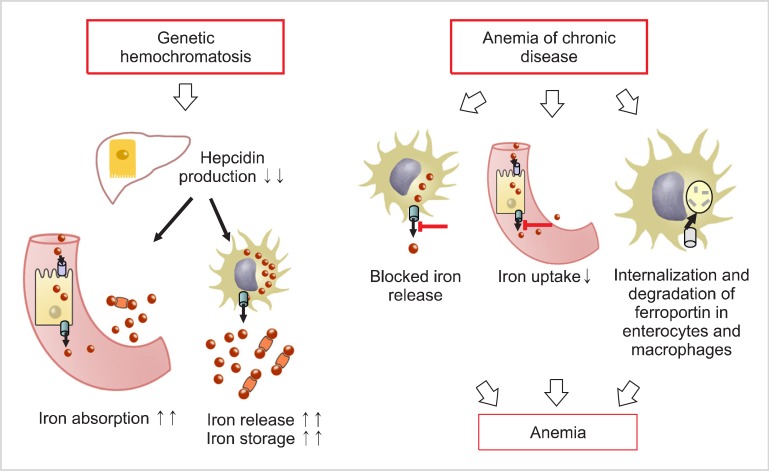
Go to :
HEPCIDIN MEASUREMENT AND RELATED PROBLEMS
Specific methods were recently developed to measure hepcidin and evaluate its diagnostic potential. The competitive ELISA test uses biotinylated or radioiodinated hepcidin as a tracer [5, 32]. However, mass spectrometry is used for the hepcidin assay; this involves standards labeled with hepcidin isotopically or standards that bind the hepcidin-truncated molecule [33, 34]. The use of different methods for the determination of hepcidin levels mainly depends on the employment of different hepcidin standards as well as the varying abilities of different methods to detect the hepcidin-20 and hepcidin-22 isoforms in addition to the hepcidin-25 bioactive isoform. Despite the varying abilities of the methods mentioned above, there is no difference between the samples and analytical methods used. This indicates that all methods are appropriate for detecting hepcidin isoforms in samples. The results of the first international round-robin research on the quantification of plasma and urinary hepcidin assays led to the conclusion that the differences in the levels of hepcidin isoforms between methods could be due to the use of different calibrators with assigned levels obtained by different techniques, hepcidin aggregation of either the standard solution or sample, binding of hepcidin to α2-macroglobulin or albumin, or existence of 3 hepcidin isoforms (hepcidin-25, hepcidin-22, and hepcidin-20) [35].
Since approximately 90% of circulating hepcidin is linked to α2-macroglobulin, it is important to consider whether it is necessary to measure total or unbound hepcidin and which current valuation methods can be used. The difference between immunochemical (IC) methods and mass spectrometry is another important issue. IC methods cannot selectively distinguish hepcidin-22 and hepcidin-25 from hepcidin-20. However, the reasons for including hepcidin-20 and hepcidin-22 in the total hepcidin level for determining different iron-related diseases remain unclear. The coefficient of variation among samples is lower for the plasma hepcidin assay than that for urinary hepcidin. This suggests that the difference between measured levels in the urine and plasma is not due to the method used, but due to biological mechanisms such as excretion. Hepcidin levels measured using different methods vary considerably, but the analytical variance is low and similar for other methods in general.
In order to harmonize the various methods used to measure hepcidin, it is recommended to introduce internal standards for all basic mass spectrometry methods used for clinical trials, reach assigned consensus levels, adjust the levels of calibrators used in each procedure, produce a calibrator that mimics a patient's serum, periodically analyze shared samples and/or interchangeable calibrators, and have a value assigned for quality control.
In addition to measuring hepcidin, it is possible to measure prohepcidin. However, the measurement of the latter does not appear to be biologically relevant with respect to hepcidin.
Another problem is the fluctuation of diurnal hepcidin values. Hepcidin levels are lower in the morning and increase in the afternoon. In addition, assay sensitivity is related to the amount of iron introduced by the diet [5, 36]. Various types of anemia exhibit different iron-related parameters including hepcidin, transferrin saturation, ferritin, and soluble transferrin receptor (Table 1).

Go to :
POSSIBLE THERAPEUTIC APPLICATION OF HEPCIDIN AGONISTS
Having established the central role of hepcidin in iron homeostasis, it is possible to consider its use as a novel treatment for anemia. Under particular conditions of chronic inflammation, despite low plasma iron levels, iron treatment can be harmful because it can trigger the growth of pathogens and/or contribute to neoplastic transformation because of its ability to unbalance immune functions, particularly the immune-mediated activities of T-cells [37, 38].
Although hepcidin is not used for therapeutic purposes at present, there are some ongoing studies on selected patients. Hepcidin agonists could be used to prevent or improve the accumulation of iron in both transfused and non-transfused β-thalassemic patients and even in anemia with iron storage. Hepcidin antagonists could be used in patients with diseases that cause hepcidin excess and occur with a framework of IDA or systemic IDA.
Other drugs targeting the regulation of the hepcidin system, such as dorsomorphin, a small molecule that inhibits the BMP signal, have been tested. Even soluble HJV acts as a BMP signal antagonist, decreasing the basal concentration of hepcidin in mice while increasing the liver iron concentration [39]. The use of anticytokines such as anti-IL-6 has been shown to suppress hepcidin production activity and improve anemia in some cases [40, 41]. Hydroxyproline inhibitors may be effective for inducing hepcidin inhibition; such inhibition could be caused not only by erythropoiesis stimulation but also by the ability of hydroxyproline inhibitors to interfere with the transcriptional complex, hypoxia-inducible factor [42].
Go to :
CONCLUSIONS
Iron-related diseases are common and clinically important. Recent research on both hepcidin and the key role of the hepcidin-ferroportin axis has clarified the pathogenesis of IDA, ACD, and anemia with iron overload. This knowledge will allow the development of new tests and possibly new treatments as well.
Go to :
 XML Download
XML Download