

 PDF
PDF ePub
ePub Citation
Citation Print
Print



I. Introduction
Benign lymphoid hyperplasia (pseudolymphoma) has been reported in the skin, lungs, orbit, and gastrointestinal tract, but only rarely in soft tissues1. Cutaneous pseudolymphoma is a term used to describe a skin lesion that bears a clinical or histological resemblance to lymphoma. Many entities are included under this umbrella based on their lymphoid composition, pattern of infiltration, and associated findings, including cutaneous lymphoid hyperplasia (CLH), pseudolymphomatous folliculitis, Kimura disease, Castleman disease, and pseudo-mycosis fungoides. CLH has had a wide variety of names, including Spiegler-Fendt sarcoid, lymphocytoma cutis, and lymphadenosis benigna cutis. It has a worldwide distribution and affects all races and ethnic groups equally. It occurs both in adults and children, with a slight female predominance 2. Due to this vast array of characteristics, the lesion is most often not included in the differential diagnosis of soft to firm lumps present on the head or neck. A systematic approach to the workup and diagnosis of such distinct lesions is discussed in this article.
II. Case Report
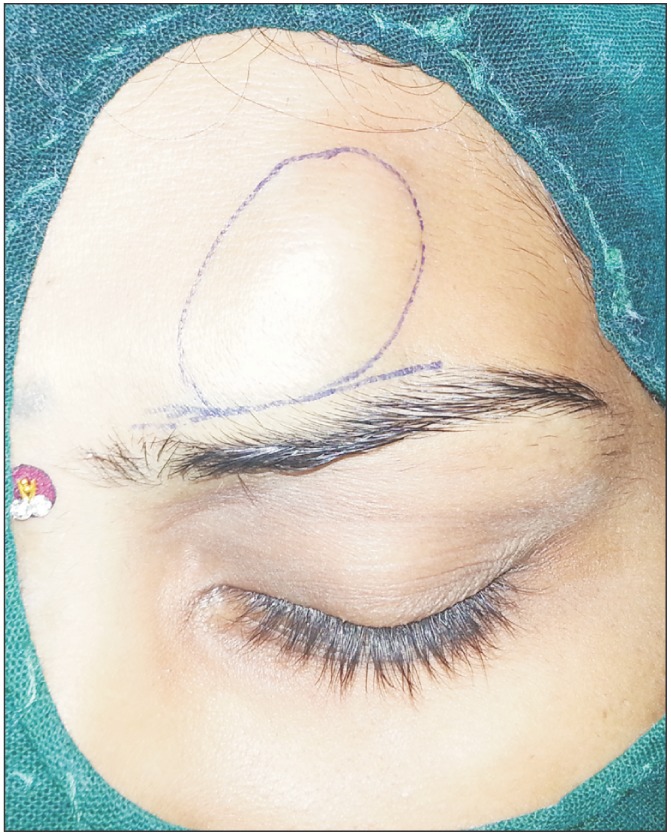
A 20-year-old Asian Indian female presented to our Oral and Maxillofacial unit with a lump on the left side of her forehead. The lump has been present for 1 month, and she had developed a psychological complex due to the unaesthetic appearance. The lesion was initially 1×1 cm2 and had eventually progressed to the presentation size over the month. There was no history of associated fever, previous treatments, unexplained weight loss, or malaise. The patient had no deleterious habits, and the family history was inconclusive for any hereditary or genetic correlation.
General examination did not reveal any significant findings such as similar lesions on the extremities, chest, or abdomen. On further local examination, a 2.5-cm-long and 3-cm-wide, well circumscribed swelling was seen over the left para median region of the forehead, which was firm to doughy in consistency and non-tender. The skin over the swelling was normal and did not show any signs of inflammation or follicular change. The swelling had not displaced the skin below the eyebrows and was not fixed to underlying structures.(Fig. 1) No neurological signs were present such as paresthesia or hyperesthesia. No regional or distant lymph nodes were palpable. Routine blood and urine investigations were ordered and were within the normal limits. Based on the clinical features, a provisional diagnosis of a solitary cutaneous neurofibroma over the left paramedian forehead was made. The differential diagnosis also included a myxoma and schwannoma of the regional neural bundle, all of which required a similar line of initial treatment. The treatment plan included an excisional biopsy due to the perceived benign nature of the lesion. The patient underwent an excisional biopsy after informed consent was obtained.
A horizontal incision was marked in a natural skin crease above the left eye brow, taking care to avoid the hair follicles. An incision was given identifying the layers till the capsule of the lesion. Supracapsular blunt dissection commenced, and it was noted that the lesion was encasing the supraorbital nerve bundle.(Fig. 2) On further circumferential dissection, the lesion was freed from the nerve sheath, which was found to be clinically intact. Complete excision of the lesion was achieved; following hemostasis, closure was performed in layers.
The histopathological processing showed a highly cellular tissue composed of dense aggregates of lymphocytes with germinal center formation interspersed with abundant mast cells in a scanty stroma. The stroma was composed of loose to dense bundles of collagen fibers forming septae separating the lesional tissue into lobules and forming a capsule. Cut and longitudinal sections of nerve bundles were also seen. Endothelial-lined blood vessels with red blood cells (RBC) and extravasated RBC were noted. These features were suggestive of lymphoid hyperplasia.
The patient underwent regular follow-up for 10 months, and no signs of recurrence, hypoesthesia, or paresthesia were noted.
III. Discussion
1. Etiology and pathogenesis
Most of the commonly diagnosed CLH are thought to be due to idiopathic etiology; however, some may be associated with bites or stings from arthropods, infections such as herpes zoster and Borrelia burgdorferi, recent tattoos, vaccinations, and use of gold jewelry2. Medications that can be associated with CLH include phenytoin (hydatoin-associated pseudolymphoma syndrome), carbamazepine, phenobarbitols, cyclosporine, calcium channel blockers, and H1 and H2 antagonists. Due to this variety of causative agents, the identification of the etiologic factors of this lesion can be considered a very enigmatic dilemma; our case was deemed to be idiopathic. It has been proposed that rather than being the target of the immune response; these agents alter the lymphocyte reactivity in a way to promote a typical CLH response.
2. Clinical findings
Clinical history taking should include the symptomology
and duration of the lesion as well as the nature and pace of progression. CLHs are usually slow growing lesions, mostly painless, and have a slow pace of progression with an indefinite rate of growth. The history should also include the so called lymphoma B symptoms such as fever of unknown origin, unexplained weight loss, night sweats, fatigue, and malaise. Our patient did not have any of these symptoms, further lessening the probability of this diagnosis. A general examination should be part of the routine analysis for cutaneous lesions in order to rule out the distribution of skin lesions and peripheral lymph nodes. Extracutaneous involvement should be ruled out by complete blood count, general chemistry panel, and chest radiographs. These lesions are usually solitary but may occur as papules, plaques, or nodules. They usually have a firm to doughy consistency and range from red-brown to violet in color. Use of topical and systemic corticosteroids should be discontinued for at least 4 weeks before biopsy as these agents attenuate the lymphoid infiltrates. The most diagnostic area of biopsy in these lesions is the deepest portion. In our case, i.e., a solitary lesion, it was best to perform an excisional biopsy as the inflammatory changes associated with an incisional biopsy would change the local environment. If uncertainty remains regarding the diagnosis between a pseudo and true lymphoma, further tests may be ordered, such as computed tomography of the chest and abdomen and bone marrow biopsies.
3. Differential diagnosis
The differential diagnoses for an acquired, long standing, painless, firm to doughy lesion occurring on the forehead are numerous, ranging from the benign solitary neurofibroma to malignant lymphomas. The key diagnoses to include are neurofibroma, lipoma, drug eruptions, infectious and inflammatory granulomas, and metastatic carcinoma. It is important to always rule out cutaneous B-cell lymphoma (CBCL) and leukemia cutis, as they are aggressive lesions, and treatment can be completely tangential.
4. Clinical course and prognosis
These lesions can resolve spontaneously, if the etiologic agents are removed, or might persist indefinitely, giving rise to a poor aesthetic outcome. Nodular scabies is a well-known clinical form of persistent CLH that presents as pruritic, persistent nodules for months even after specific treatment. Single standing lesions usually resolve after excisional biopsy. One of the most challenging outcomes is the close relationship with CBCL. Most patients subsequently diagnosed with CBCL receive a previous diagnosis of CLH based on biopsy results. Patients with papulosis, granulomatosis, and lymphadenopathy have a high chance of developing lymphomas 2. Some authors present this as a “concept of continuum”, where the cutaneous pseudolymphomas, such as CLH, represent the benign end of the lymphoproliferative continuum, with the intermediate form being clonal CLH, and the malignant extreme being true lymphomas. This concept is important when considering treatment options based on stage in the continuum, and regular follow up is needed due to the increased risk of clonally related lymphoma and the other lymphoid abnormalities usually associated with this lesion3.
5. Treatment
CLH can be treated in a variety of ways based on the etiology. Lymphocytoma cutis, traditionally caused by B. burgdorferi, responds to cyclosporin therapy, as do some of the idiopathic varieties. Glucocorticosteriods (topical, intra lesional 5-40 mg/mL 1 mL monthly or systemic 60/40/20 mg 5 days each tapered), hydroxychloroquine 200 mg twice daily, and minocycline 50-100 mg twice daily have been used in the past with varied success4. Some studies also support the use of HIV antiviral triple therapy5 or topical tacrolimus6. Radiotherapy has been shown to be effective but is mostly considered a last resort. Recently, the use of pulsed dye lasers and photodynamic therapy has been shown to be effective in esthetic areas7. Our case was an atypical CLH; in most instances, these lesions cannot be distinguished from true lymphoma, and was hence treated as a localized lymphoma, i.e., excision, after the staging ruled out extra cutaneous disease. Due to the local and distant recurrences after any of the above treatments, this may have been followed by radiotherapy; this was not so in our case due to the patient's desires.
6. Prevention
Key to the prevention strategy of CLH is to avoid the ingestion of, or contact with, any of the causative agents in patients with a suggestive clinical history, such as anticonvulsants, arthropods, gold jewelry, and hair dyes8.
Due to the overlapping clinical and histological characteristics of CLH with many other lesions, it is important to include and exclude this lesion in the differential diagnoses put forward for suspected cutaneous lesions.

 XML Download
XML Download