

 PDF
PDF ePub
ePub Citation
Citation Print
Print



I. Introduction
Approximately 20% of head and neck cancers occur in the oral cavity in Korean populations. Squamous cell carcinoma (SCC) is the most frequent oral cancer, accounting for more than 90% of all intraoral malignant tumors.
Despite technological advancements in the fields of surgery, radiotherapy and chemotherapy, the prognosis of locally advanced oral SCC (OSCC) patients remains poor, with only a 50% survival rate over 5 years1. Because of the poor survival rates and severe functional impairments caused by surgery and radiation, it is critical to develop novel therapeutic strategies for the treatment of advanced OSCC.
Cisplatin, a potent chemotherapeutic agent, has been shown to have significant clinical activity against various solid tumors, and it has been accepted as the standard regimen for various cancers treatments including OSCC. The main mechanisms of cisplatin cytotoxicity involve cisplatin interaction with DNA to form unique DNA adducts2. These DNA adducts further modulate the expression of several signal transduction pathways involved in cell survival, including p53.
The major problems with cisplatin application are drug resistance and toxic side effects, which lead to discontinuation or limited therapeutic efficacy3. To overcome these problems, combination therapy with other agents is recommended. The identification of the mechanism underlying the cisplatin effect will inform the selection of appropriate agents that can activate alternate pathways to induce a synergistic effect as well as reduce resistance and cytotoxicity4.
The activation of p53 is regarded as the major effect of cisplatin treatment5. p53 interacts with many key transcription factors related to several important biological functions, such as DNA repair, cell cycle regulation and apoptosis. Thus, p53 plays an important role in cancer onset and progression67.
The p53 gene mutation is detected in more than 50% of OSCC cases. Therefore, it is important to assess the relationship between cisplatin resistance and p53 mutation status. The influence of p53 on the anti-cancer effect of cisplatin in OSCC cell lines showing different p53 mutation status also needs to be investigated.
The purpose of this study was to evaluate the anti-cancer activity of cisplatin by studying the effect of cisplatin on cell viability and the mechanisms underlying the induction of cell cycle arrest and apoptosis in OSCC cell lines with genetically different p53 mutation statuses.
II. Materials and Methods
1. Reagents
Cisplatin (Dong-A Pharm., Seoul, Korea) was dissolved in distilled water at 10 µg/mL, which is the highest concentration used in vitro, and stored in aliquots at −20℃ until use. Final concentrations between 0.5 and 10.0 µg/mL of cisplatin were obtained by appropriate dilutions of the stock compound with the defined medium.
2. Cell lines and cell culture
Three OSCC cell lines, YD-8 cell line (p53 point mutation, tongue), YD-9 cell line (p53 wild type, buccal cheek), and YD-38 cell line (p53 deletion, lower gingiva) were used8. All cell lines were purchased from the Korean Cell Line Bank (Seoul, Korea).
Each cell line was maintained in RPMI-1640 medium (Gibco, Grand Island, NY, USA) supplemented with 10% heat-inactivated fetal bovine serum (FBS; Gibco), 100 µg/mL of streptomycin and 100 IU/mL of penicillin (Gibco) as a monolayer under standard conditions (37℃, 5% CO2, humidified atmosphere). To transfer or passage the cell lines, each confluent monolayer was washed with phosphate-buffered saline (PBS; Welgene, Gyeongsan, Korea) and detached with a 0.05% trypsin/0.02% ethylenediaminetetraacetic acid solution (Gibco).
3. MTS viability assay
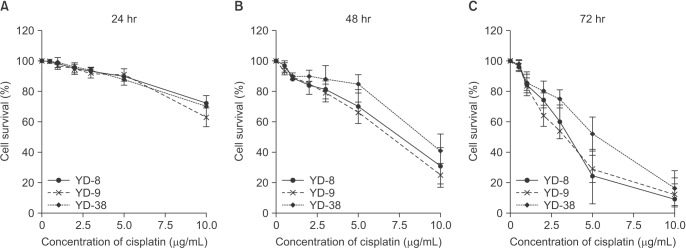
Cells at a density of 2×104 were added to a 96-well plate in 100 µL of RPMI with 10% FBS. The control samples of each cell line were treated with medium only. Cells were treated with different concentrations (between 0.5 and 10.0 µg/mL) of cisplatin for 24, 48, and 72 hours. For the viability assay, 20 µL/well of CellTiter 96 AQueous One Solution Reagent (MTS; Promega, Madison, WI, USA) was added. After 1 hour under standard conditions, the absorbance at 490 nm was recorded using an ELISA plate reader. The experiments were performed in triplicate with three independent experiments for each condition. The data (±standard error) were normalized to the control samples. The 50% inhibition concentration of cell viability (IC50) value was calculated from the dose-response curve.
4. Cell cycle analysis
The cells were plated at a density of 1×106/well in 100 mm culture dishes. The cells were treated with 0.5 and 1.0 µg/mL of cisplatin for 48 hours, harvested by centrifugation at 1,200 rpm for 3 minutes, washed twice in ice-cold PBS, fixed in 70% ethanol, stored at −20℃ for a minimum of 1 hour, washed with ice-cold PBS and then resuspended in 500 µL of propidium iodide (PI)/RNase Staining Buffer (BD Biosciences, San Jose, CA, USA). The cell cycle position was evaluated by Becton-Dickinson Fluorescence-Activated Cell Sorting (FACScan) using an excitation laser set at 480 nm and a detection wavelength of 575 nm (Becton-Dickinson, Mountain View, CA, USA). A minimum of 10,000 events/sample were analyzed.
5. Annexin assay
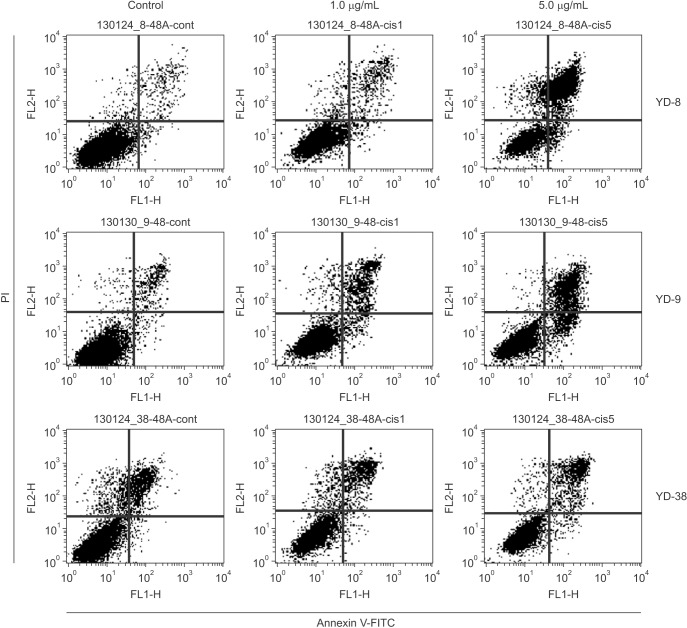
Apoptosis was quantified using the FITC-Annexin V Apoptosis Detection Kit I (BD Biosciences) according to the manufacturer's instructions. Briefly, the cells were plated at a density of 1×106/well in a 100 mm culture dish. The cells were treated with 1.0 and 5.0 µg/mL of cisplatin for 48 hours and harvested by centrifugation at 1,200 rpm for 3 minutes. The cell pellets were resuspended in annexin V binding buffer (150 mmol/L NaCl, 18 mmol/L CaCl2, 10 mmol/L HEPES, 5 mmol/L KCl, 1 mmol/L MgCl2). FITC-conjugated annexin V (1 µM/mL) and PI (50 µM/mL) were added to the cells and incubated at room temperature in the dark for 15 minutes. The analyses were performed using a FACScan instrument (Becton-Dickinson). Annexin V-positive cells were regarded as apoptotic.
6. Western blot analysis
Cisplatin-treated and untreated cells were suspended in RIPA buffer (Rockland, Gilbertsville, PA, USA) containing 5 µM of AEBSF, 1.5 nM aprotinin 10 nM E-64, 10 nM leupeptin, sodium orthovanadate, sodium molybdate, sodium tartrate and imidazole, and were placed on ice for 20 minutes. The supernatant was collected after centrifugation at 4℃ at 13,000 rpm for 20 minutes. The protein concentration was determined using a BCA Protein Assay Kit (Pierce, Rockford, IL, USA). The whole lysate (20 µg) was resolved on 10% or 12.5% SDS-PAGE gel, transferred onto a PVDF membrane (Bio-Rad, Hercules, CA, USA) by electroblotting and probed with human anti-β actin, anti-p53, anti-p21, anti-caspase 3, anti-caspase 7, anti-caspase 9, or anti-PARP. The membrane was washed with PBS-T (1×PBS and 0.05% Tween-20) and incubated for 1 hour with an HRP-conjugated anti-rabbit or anti-mouse antibody. The blot was developed using an enhanced chemiluminescence kit (iNtRON Biotech, Seongnam, Korea).
III. Results
2. Cisplatin induces cell cycle arrest at the G2/M phase
Cell cycle analysis was performed after treatment with 0.5 and 1.0 µg/mL cisplatin for 48 hours in each cell line. Cisplatin induced G2/M cell accumulation in all cell lines.(Fig. 2) The cell population of the G2/M phase increased from 11.46%±3.22% to 52.87%±11.66% for the YD-8 cell line, from 30.66%±2.42% to 64.32%±0.49% for the YD-9 cell line and from 24.52%±8.47% to 58.26%±9.65% for the YD-38 cell line after treatment with 0.5 µg/mL cisplatin. The cell population of the G1 phase decreased from 80.28%±5.43% to 30.19%±4.57% for the YD-8 cell line, from 46.41%±8.21% to 14.24%±9.31% for the YD-9 cell line and from 56.89%±7.99% to 22.24%±9.44% for the YD-38 cell line after treatment with 1.0 µg/mL cisplatin.
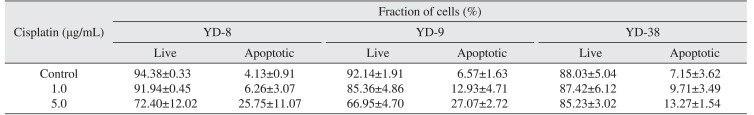
3. Induction of apoptosis by cisplatin in OSCC cell lines
This staining method along with flow cytometry enables the quantitative assessment of living (annexin V-FITC negative/PI negative), early apoptotic (annexin V-FITC positive/PI negative), late apoptotic (annexin V-FITC positive/PI positive) and dead/necrotic (annexin V-FITC negative/PI positive) cells. The effects of 48-hour cisplatin treatment on YD cell apoptosis are shown in Fig. 3 and Table 2. The cells in the lower right quadrant represent early apoptosis, and the cells in the upper right quadrant represent late apoptosis. The proportion of cells staining positive for annexin V-FITC increased in a dose-dependent manner following cisplatin treatment. The apoptotic proportion was lower in the YD-38 cell line compared to the YD-9 and YD-8 cell lines.
4. Cell cycle alteration or apoptosis-related protein expression induced by cisplatin
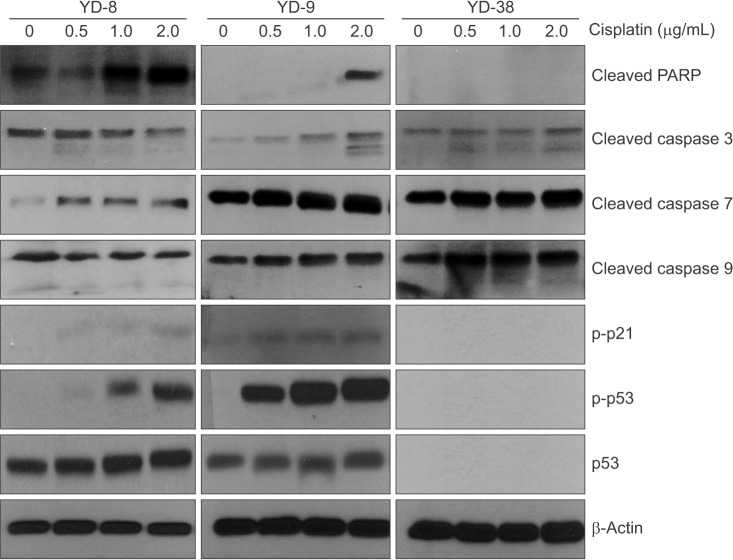
Since cisplatin induced G2/M arrest in the OSCC cell lines, we expected to see alterations in the cell cycle regulatory proteins after treatment with a non-cytotoxic doses of cisplatin (0.5, 1.0, and 2.0 µg/mL). In the cell cycle, distinct cyclin/CDK complexes are activated to regulate cell cycle progression 91011. With respect to p53-dependent cell cycle alterations, p21 (a p53-regulated cyclin dependent kinase) is considered a critical component12. Levels of p53 protein were detected by immunoblotting at 48 hours in the YD-8 and YD-9 cell lines, but not in the YD-38 cell line.(Fig. 4) Also, immunoblotting analysis showed that the phosphorylation of p21 increased in the cisplatin treatment only in the YD-8 and YD-9 cell lines.
In the evaluation of apoptosis, as shown in Fig. 4, caspase 3, caspase 7, caspase 9 and PARP protein expression was increased at 48 hours in all cell lines after cisplatin treatment compared with the controls.
IV. Discussion
Cisplatin has been widely used for the treatment of a variety of solid tumors including OSCC and for the evaluation of the resistance mechanism and the effect of combination therapy61314. Although the mechanism of action for cisplatin is not fully understood, it is thought that the effects of cisplatin are partly related to p53, a tumor suppressor gene, which plays an important role in cancer. Because more than half of all human cancers lose p53 function through mutations, it is important to study the potential impact of p53 mutations on disease pathology and therapeutic responses. Cancers with an inactive, mutant p53 are aggressive and are often resistant to ionizing radiation and chemotherapy. However, there are conflicting data concerning the influence of the p53 gene on the efficacy of chemotherapeutic agents including cisplatin. Weinstein et al.15 reported that cell lines with p53 mutations are generally more resistant to DNA-damaging agents than cell lines with wild-type p53 from in vitro study of various cell lines. Clinically, poor prognosis of tumors with p53 mutations is expected with various types of tumors161718. In another study, however, the cytotoxic effect of cisplatin was more severe in mutant p53 cell lines than in wild-type p53 cell lines19. Many researchers suggested that there are p53-dependent and p53-independent pathways, respectively202122.
Cell lines are useful for understanding the mechanism of action and resistance of chemotherapeutic agents. However, it has been reported that the success rate of OSCC cell line establishment is very low. Also, genetic abnormalities of human cancer are geographically-dependent, so cultural and environmental backgrounds are closely related to the carcinogenic process8. In this study, the YD-8, YD-9, and YD-38 cell lines, which originated from Korean oral cancer patients, were used8; these three cell lines were derived from untreated primary tumors of the tongue (YD-8 cell line), buccal cheek (YD-9 cell line), and lower gingiva (YD-38 cell line), respectively, and have genetically different p53 statuses. The YD-8 cell line has a point mutation at codon 273 of exon 8, changing an arginine to histidine in the DNA-binding site, revealing a key role for p53 transcriptional activation. The R273H mutation accounts for approximately 20% of reported p53 missense mutations23. The YD-9 cell line does not have the p53 mutation, and p53 protein is positively expressed in the YD-9 cell line. The YD-38 cell line has a p53 deletion and the p53 protein is not expressed in the YD-38 cell line. Because p53 mutation is common in OSCC, evaluation of cell cycle arrest and apoptosis in OSCC cell lines with various p53 statuses could be informative for developing cancer therapy.
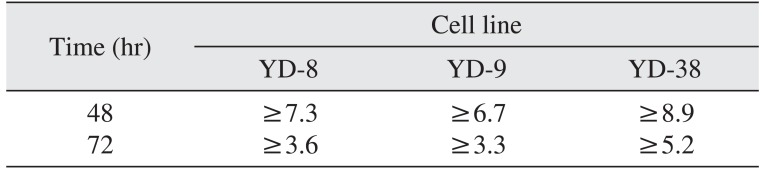
In the present study, cisplatin showed cytotoxicity via G2/M phase arrest and apoptosis in OSCC cell lines in dose- and time-dependent manner. The cytotoxic effect of cisplatin was more prominent in the YD-9 cell line with wild-type p53 and the YD-8 cell line with p53 point mutation than in the YD-38 cell line with p53 deletion.(Fig. 1, Table 1) The IC50 value was also relatively lower in the YD-9 and YD-8 cell lines than in the YD-38 cell line.
Interestingly, after the application of cisplatin, p53 protein was detected in the YD-8 and YD-9 cell lines via immunoblotting.(Fig. 4) There are several possible mechanisms of p53 mutation that can result in the overexpression of the p53 protein. Furthermore, mutations in the promoter or in the intron of the p53 gene could result in a higher expression of the wild-type p532425. In the YD-8 cell line with a p53 point mutation, the abnormal p53 protein might be detected by immunoblotting due to a prolonged half-life. Also, we suspect that p53 expression status at the protein level is more important than p53 mutation status because the YD-9 and YD-8 cell lines showed similar responses to cisplatin treatment despite the difference in p53 mutation status.
In relation to cell cycle arrest, p21 was detected in the YD-8 and YD-9 cell lines by immunoblotting. It is generally accepted that p21, a p53-regulated cyclin-dependent kinase, is involved in the cell cycle alteration caused by DNA damaging agents12. Cell cycle arrest related to p53 was expected to be blocked in the YD-38 cell line because p53 and p21 were not detected in the YD-38 cell line via immunoblotting. However, cell cycle arrest at the G2/M phase, a decrease in the percentage of cells in the G1 phase and an increase in the percentage of cells in the G2/M phase, were detected after treatment of the cells with cisplatin for 48 h in all three cell lines in this study.(Fig. 2) The cell cycle alteration effect of cisplatin was not associated with the expression of p53 or p21, which indicates that p53- and p21-independent pathways are involved in cell cycle arrest in the cell lines used in this study.
Also, more apoptosis was detected in the YD-8 and YD-9 cell lines than in the YD-38 cell line after cisplatin treatment. (Fig. 3, Table 2) Although the p53-dependent apoptotic pathway might be blocked in the YD-38 cell line, caspases and PARP were detected in the YD-38 cell line by immunoblotting. However, the YD-38 cell line showed less apoptosis in FACS analysis in this study, suggesting that deletion of p53 in the YD-38 cell line is related to apoptosis.
V. Conclusion
The results of this study clearly indicate that cisplatin has in vitro anti-cancer effects against OSCC cell lines via G2/M phase arrest and apoptosis. However, there were also some differences in the cytotoxic effect of cisplatin among OSCC cell lines in this study. Multiple factors might contribute to these differences, such as the tumor entity or tumor characteristics. One factor may be mutation status, which could play a critical role in carcinogenesis. For this study, we focused on p53 as a possible candidate and found that p53 status of the patients should be considered before choosing cisplatin chemotherapy. Further studies on p53 mutation status are necessary to understand the biological behavior and characteristics of OSCC and consequently to establish appropriate treatment.

 XML Download
XML Download