

 PDF
PDF ePub
ePub Citation
Citation Print
Print



I. Introduction
Oral cancer is generally classified as head and neck cancer, and head and neck squamous cell carcinoma (HNSCC) is the sixth to ninth most common malignancy in the world12. Some authors have classified oral cancer as oropharyngeal cancer (OPC), and oropharyngeal squamous cell carcinoma (OPSCC) has been confused with oral squamous cell carcinoma (OSCC). In our previous report on the incidence of oral cancer in Korea1, we identified oral and maxillofacial cancer according to anatomic site, dividing it into 10 areas of the lip, tongue, mouth, salivary glands, tonsil, oropharynx, nasopharynx, hypopharynx, pharynx unspecified, and nose and sinuses.
Cervical cancer is the second most frequent cancer in women worldwide, and human papilloma virus (HPV) infection causes most cervical cancers and a variable proportion of certain non-cervical malignancies, including vulvar, vaginal, penile, anal, and OPC34. More than 150 different subtypes of HPV have been reported globally, and types 16 and 18 are associated with the onset of 70% of cervical cancers worldwide567. Smoking and alcohol are well known risk factors of oral cancer1, as is HPV infection, and the relationships between these known factors warrant closer examination.
In this article, we focus only on oral cancer with mucosal immunity and multistage carcinogenesis characteristics related to virus infection. We review current HPV-related vaccination recommendations and recent research on oral cancer vaccination based on the specific subtypes of HPV found in oral cancer patients.
Go to : 
II. HPV and Its Pathogenesis in Cancer
1. HPV structure and classification
HPV is part of an ancient family of pathogens known to infect epithelial tissues of amphibians, reptiles, birds, and mammals. This group of DNA viruses forms a separate Papovaviridae family, which includes Papillomaviridae and polyomaviruses78.
HPV is formed by a non-enveloped icosahedral capsid with circular double-stranded DNA. This small genome is comprised of 8,000 base pairs with three distinct regions: the early region (E), late region (L), and upstream regulatory region (URR) or long control region. The URR is found between the E and L regions and contains promoter and enhancer DNA sequences that are critical for regulating viral replication and transcription of both viral and cellular genes789. The URR I is a non-coding region, but the E and L regions are coding regions with open reading frames (ORFs). The E region contains seven to eight genes; E1 functions in viral replication by initiating DNA replication and transcription, E2 regulates viral transcription and DNA replication by controlling ORFs E6 to E7, E4 interacts with cytoskeleton proteins by altering the extracellular matrix cell, E5 interacts with cellular proteins and downregulates major histocompatibility complex class 1 molecules, E6 degrades p53 oncoproteins, and E7 binds to the Rb oncoprotein. The functions of E3 and E8 are still poorly understood, with the exception of a few HPV subtypes8910. The L region contains the L1 major viral capsid structural protein and L2 minor viral capsid structural protein, which encode structural proteins that are necessary for viral capsid formation in the final stages of replication.
HPV can be phylogenetically classified into genera, species, and types. Genera classification includes alpha, beta, gamma, mu, and nu. More than 150 types of HPV are currently known, and approximately 120 types are fully sequenced10. The classification of HPV types is based mainly on sequence analyses of the L1 gene, which is the most conserved gene in all known papilloma viruses. HPV types can be differentiated by less than 10% homology in the L1 ORF, subtypes by less than 2% to 10% homology, and variants by less than 2% homology (2% in coding regions and 5% in non-coding regions)811. Approximately 80% to 90% of similarities in the same species share biological properties such as tissue tropism, disease manifestation, and pathogenicity.
HPV is also classified according to cutaneous or mucosal tropism characteristics. The cutaneous types are associated with skin lesions; HPV 1, 2, and 4 are most prevalent in plantar warts, and HPV 5, 8, 9, 12, 14, 15, 17, 19-25, 36, 46, and 47 are frequently found in epidermodysplasia verruciforme. HPV 5 and 8 are also related to skin carcinomas121314. In contrast, mucosal types infect the anogenital tract and upper aerodigestive tract and include HNSCC, OPSCC, and oral cancer. Mucosal types can be subdivided into low-risk and high-risk types based on oncogenic potential. The most relevant low-risk types are HPV 6 and 11, and HPV 40, 42, 43, 44, 54, 61, 70, and 72 can be observed in benign genital mucosal lesions. HPV 31, 33, 35, 52, 58, and 67 are known to be moderate to high-risk, and among the high-risk types, HPV 16 and 18 are most common, and type 16 can be found in various cancers such as cervical cancer, OPSCC, and penile carcinoma12151617.
2. General pathogenesis in cancer
The HPV life cycle is initiated through microlesions in the epithelium and is characterized by a specialized differentiation program of infected squamous epithelial keratinocytes, where viral DNA synthesis and expression are linked to capsid proteins. Virions released from the stratum corneum and granulosum directly infect the basal layer through capsid synthesis and late and early promoter activation.(Fig. 1) The HPV receptors that are involved have not been fully identified, but research has indicated that the α6 integrin with heparin sulfate in keratin-filled sacs is a possibility.
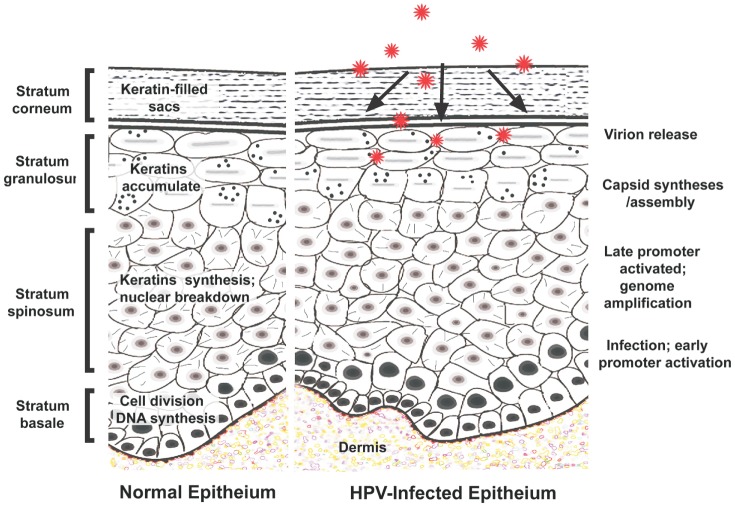
Three major replication mechanisms occur after virion penetration: plasmid, vegetative, and productive replication18. Infection usually starts in the basal and para-basal cells of the squamous epithelium. Changes in the keratinocytes from the basal layer to the surface of the epithelium provide a suitable micro-environment for productive cell replication, responsible for transformation of the keratinocyte into a permissive cell18. Plasmid replication occurs in the lower epithelium and can be subdivided into the amplification stage of viral DNA of up to 50 to 400 couples/diploid genomes and the maintenance stage of a constant number of couples for several cell generations. Normal viral replication is a highly regulated process, depending both on select viral proteins codified by the viral genome and on the degree of infected cell differentiation1819.
Vegetative replication, which occurs in cells that differentiate from the epithelium, involves a link between cell differentiation and viral expression of the gene. To activate infection, the virus must have access to the ‘generative’ compartment of the epithelium through exposure of superficial layers in order to reach the basal layer, where the specific α6 integrin receptor is present1819. During the initial phase of infection, when the virus colonizes basal and para-basal cells of the epithelium, the viral genome undergoes episomal replication, since it is present as an extra-chromosomal fragment of circular DNA. At this stage of episomal or early replication, relatively few copies of viral DNA (20-200) are present per host cell. The episomal form acts as a reservoir of infected cells, which are morphologically indistinguishable from non-infected cells, and is responsible for the latent status of infection1820.
When the infection becomes productive, the viral genes are expressed sequentially from early to late genes, following epithelial squamous differentiation, starting from basal and para-basal cells, where early portions of the viral genomes are more active, and proceeding to higher epithelial layers of both the intermediate and superficial layers, along with complete virion formation1821. This completes productive replication, in which the virus is expelled from the epithelial cells on desquamation and transmitted by direct or indirect contact.(Fig. 1)
HPV integration occurs through viral episomal DNA rupture, in conjunction with E1 and E2 ORFs, with preservation of E6 and E7 segments that may therefore undergo transcription22, causing a disturbance in cell control mechanisms and increased proliferation of infected cells. The consequence is unregulated cellular replication with an increased risk of chromosomal aberrations and excessive production of viral proteins that interact with cellular proteins2324.(Fig. 2)
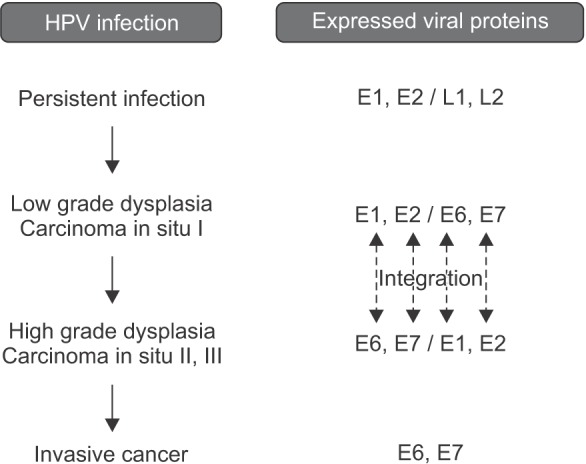
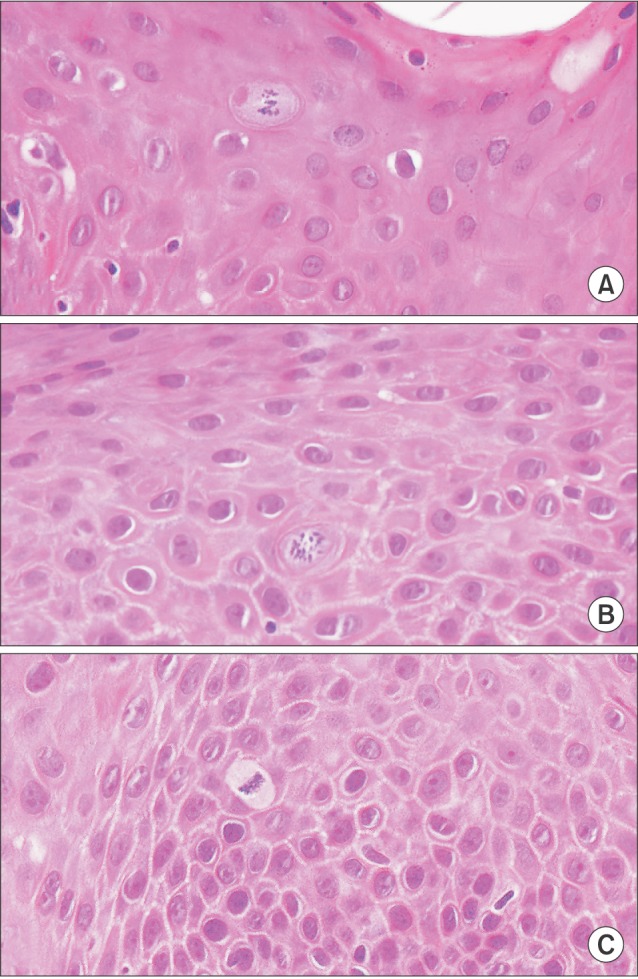
Classic viral cytopathic effects can manifest at this stage. Upon microscopic examination, HPV infection can appear as acanthosis, dyskeratosis, keratinocyte multinucleation, and koilocytosis. In particular, koilocytosis is considered to be evidence of a viral cytopathic effect, where cells show a thicker cytoplasm at the cellular membrane level and an atypical morphologically collapsed and stellate nucleus2526. A few mitosoid cells may be present among the normal keratinocytes, and chromatin peripheralization with inclusion bodies has been observed in the stratum granulosum, spinosum, and basale (Fig. 3) with suspected viral infection27. The viralinduced cell growth mechanism is analogous to that of other viruses in tumors that deregulate the cell cycle. The appearance of HPV-persistent cancer is related to the overexpression of E6 and E7 proteins. E6 interferes with the function of p53 and E7 with the function of the pRb protein, leading to abnormal cell growth by promoting inhibition of apoptosis and dysregulation of the cell cycle, respectively22.(Fig. 2)
3. Relationship to oral cancer
Until recently, approximately 20% of oral cancers and 60% to 80% of OPC were thought to be attributable to HPV infection. In 2012, the International Agency of Research of Cancer (IARC) declared that there was sufficient evidence to associate a subtype of HPV 16 with oral cancers28. Additionally, these HPV-related oral cancers differ from HPV-negative tumors or cancers in their clinical response and overall survival rates2930. In the oral cavity, 24 types of HPV, 1, 2, 3, 4, 6, 7, 10, 11, 13, 16, 18, 30, 31, 32, 33, 35, 45, 52, 55, 57, 59, 69, 72, and 73, have been associated with benign lesions, and 12 types, 2, 3, 6, 11, 13, 16, 18, 31, 33, 35, 52, and 57, with malignant lesions3132. A total of 99% of HPV infections in HNSCC are related to high-risk types 16, 18, 31, or 3333, with HPV 16 as the most common subtype and HPV 33 accounting for up to 10% of cases3334.
Epidemiologically, HPV-positive HNSCC occurs more frequently in younger patients (younger than 50 years), which differs from the typical age of HNSCC patients. A direct correlation between HPV-positive patients and sexual behavior has also been shown in HNSCC35. High-risk HPV-16 is correlated with vaginal or oral sex and frequent sexual encounters without barrier usage. Current changes in sexual practices, including first sexual experience at an earlier age, high number of sexual partners, and high probability of oral sex, may be associated with the increasing prevalence of HPV infection36.
Clinically, HPV-associated tumors can appear as a strawberry-like exophytic lesion, frequently at the base of the tongue or in the tonsil area. Most show poorly differentiated pathologic findings and cystic changes in the metastatic neck lymph nodes37. As described, the transformation of normal oral mucosa in OSCC could be related to precancerous lesions, such as oral leukoplakia (OL), oral erythroplakia (OE), oral lichen planus (OLP), nicotine stomatitis, tobacco pouch keratosis, and oral submucous fibrosis38. The role of HPV in malignant transformation of precancerous lesions has not been confirmed, but OL has been reported as the most frequent potentially malignant lesion39, OE can be associated with severe epithelial dysplasia combined with carcinoma in situ or invasive carcinoma40, the chronic mucocutaneous type of OLP is susceptible to HPV infection, and p16 with INK4a protein is a reliable precancerous marker in smokeless tobacco keratosis41.
Gene expression profiles also differ in HPV-positive oral cancers based on evidence from different pathways, such as the p53 and pRb pathways involved in cell cycling, the EGFR pathway, which is an important therapeutical target (especially in breast and lung cancers), the TGFβ pathway, the PI3K-PTEN-AKT pathway, and angiogenesis and hypoxia pathways42. The wide variation in HPV prevalence might result from different detection techniques, small sample numbers, epidemiologic characteristics of populations, and sampling techniques43. Sampling techniques for HPV include microscopy, ELISA, Southern blot, dot blot, hybrid capture, DNA microarray, and ligase chain reaction for probe amplification3443. Although a standard procedure has not yet been generally accepted, both polymerase chain reaction and in situ hybridization assays are well validated, and gene expression by DNA microarray has recently gained acceptance as a high throughput method.
Go to :
III. Worldwide HPV Vaccine Application
Since the introduction of the HPV vaccination in several European countries in 2007, more than 40 countries have introduced HPV vaccination into their national immunization programs (NIPs). According to each country's health care infrastructure and systems, basic NIPs slightly vary in coverage of young adolescent girls. Generally, HPV vaccination is recommended for females aged 11 to 26 years in many countries, as well as adolescent males in several countries, such as Australia4445.
1. History and current vaccination
In 1975, zur Hausen46 first discovered that HPV infection is the major causative agent of cervical cancer and concluded that HPV can play a pivotal role in human cervix carcinogenesis. In 1991, Zhou et al.47 developed essential virus-like particle (VLP) technology for HPV vaccination, in which 72 capsomers, each comprising five L1 proteins, are assembled into a VLP that exhibits a virus-like structure. This VLP has higher safety and antigenicity and contains no foreign DNA48. zur Hausen49 then identified subtypes HPV 16 and 18 in cervical cancer, confirming that HPV infection is the etiologic factor in almost 100% of cervical malignancies. After these findings, many studies have found HPV infection in different sites of the human body, including the skin, urethra, nasal cavity, paranasal sinus, larynx, tracheobronchial mucosa, and oral cavity50.
In 1983, Syrjänen et al.51 presented a possible correlation between HPV and oral lesions, including non-neoplastic, benign, and malignant lesions, using light microscopy. Confirmatory evidence of a relationship between HPV and the DNA of oral cavity lesions was presented by Cox et al.52 in 1991. An increasing body of related literature within the last 20 years suggests that HPV DNA is a possible etiologic factor of oral cancer, although the evidence is not as reliably demonstrated as the evidence in cervical cancers.
There are currently two U.S. Food and Drug Administration (FDA)-approved cervical cancer vaccines. Gardasil (Merck Co., Rahway, NJ, USA) is manufactured as a quadrivalent HPV (qHPV) vaccine, and Cervarix (GSK, Middlesex, UK) is produced by GlaxoSmithKline plc. as a bivalent HPV (biHPV) vaccine. The HPV L1 protein can efficiently self-assemble into VLPs, which are highly immunogenic. The qHPV vaccine, composed of VLPs from the viral L1 major capsid proteins of subtypes HPV 6, 11, 16, and 18 subtypes, is produced in yeast and uses aluminum hydroxyphosphate sulfate as an adjuvant. The biHPV vaccine contains the VLP form of HPV 16 and 18 L1 proteins and uses an AS04 adjuvant, which is a combination of aluminum hydroxide and monophosphoryl lipid A (MPL)53.
Cervical cancer elimination can be accelerated through herd immunity, the development of more affordable and accessible vaccines, education about HPV infections and sexually transmitted diseases, and increased availability of vaccinations in developing countries. A standard immunization schedule for HPV vaccination in developed countries has been adopted. For females, either the qHPV vaccine or biHPV vaccine is recommended in a three-dose series for routine vaccination at 11 or 12 years of age and also at 13 to 26 years of age. For males, the qHPV vaccine is recommended in a three-dose series for routine vaccination at age 11 or 12 years and also at 13 to 21 years of age, if not previously vaccinated. Males aged 22 to 26 years may also be vaccinated if there are medical, occupational, life-style, or other indications54. The US Centers for Disease Control and Prevention (CDC) recommendation for HPV immunization in adults is based on its demonstrated efficacy to prevent genital warts and anogenital cancers in randomized clinical trials55.
2. Previously developed vaccines
The development of the biHPV vaccine by GSK began in 1998, received FDA approval in 2009, and is now licensed in more than 100 countries and approved in more than 60 countries56. This biHPV vaccine includes the AS04 self-developed adjuvant system, which is composed of MPL, and activates cellular and humoral immune responses with the general adjuvant aluminum hydroxide adsorbed form57. MPL, the major constituent of AS04, is a modified diphosphoryl lipid A derived from Salmonella minnesota R595 that removes phosphate and fatty acid groups5657. The safety of the biHPV vaccine and the degree of immune response induction against monovalent/biHPV VLP types 16 and 18 were first confirmed through a phase I clinical study. A phase II clinical trial was conducted to evaluate the safety of vaccine composition and immunogenicity, followed by phase III studies to verify the efficacy of the vaccine in HPV-infected and/or uninfected populations in the following age groups: 15-25 years, 10-25 years, 10-14 years, and 15-55 years58.
The qHPV vaccine from Merck was initially developed as an HPV L1 VLP-based prophylactic vaccine in several research institutes and was discovered to have the same structure as HPV 16, which was used as a main constituent in 1993, and was approved by the FDA in 2006596061. This vaccine has been approved in 121 countries in 2011, and over 74,000,000 doses have been administered globally. Four viral antigens produced from the yeast Saccharomyces cerevisiae expression system comprise the qHPV vaccine; each gene is expressed using the yeast expression vector pGAL110, which is induced by galactose and is followed by cell harvesting6061.
Purified VLPs are adsorbed onto aluminum hydroxyphosphate sulfate and formulated with sodium chloride, sodium borate, L-histidine, polysorbate-80, and water for injection62. The antigens that are produced are combined with the adjuvant to form the qHPV vaccine, and the detailed combination includes 20 µg of subtype 6 L1 protein, 40 µg of subtype 11 L1 protein, 40 µg of subtype 16 L1 protein, 20 µg of HPV subtype 18 L1 protein, and 225 µg of aluminum hydroxyphosphate sulfate63.
3. Adverse effects of HPV vaccines
The adverse effects of biHPV and qHPV are not serious compared to those of other popular vaccines. They include fever (18%), local reaction (16%), exanthema or erythema (7%), menstruation problems (7%), headache (7%), and malaise (6%)53. According to the vaccine adverse event reporting system, there were 12,424 reported cases of adverse effects following immunizations (AEFIs) with the qHPV vaccine for women aged 9 to 26 years in the United States in 201364. This represents a 53.9/100,000 ratio of adverse effect onset. Of these cases, 772 cases (6.2%) were reported as serious AEFIs. Among the serious AEFIs from the 100,000 qHPV doses detailed, 8.2% were syncope, 7.5% local site reactions, 6.8% dizziness, 5% nausea, 4.1% headache, 3.1% hypersensitivity, 2.6% urticaria, 0.2% each of venous thromboembolic events, autoimmune disorders, or Guillain-Barre syndrome (GBS), 0.1% each of anaphylaxis and death, 0.04% transverse myelitis and pancreatitis, and 0.009% motor neuron disease. Most did not meet the FDA definition of serious side effects, including GBS64, a rare neurological disorder that induces muscle weakness. The FDA and CDC reexamined several reports and concluded that qHPV vaccination is not related to GBS onset, although further monitoring and study are needed65.
There were 647 cases of spontaneous adverse events in a mass vaccination campaign study of the biHPV vaccine for girls aged 15 to 18 years in the UK from 2009 to 2011. This resulted in a ratio of 11.6/10,000, and 61% of the events were the result of causality assessments53. Examples of generally unreported cases include 5 cases of complicated migraines and single cases of GBS, Bell's palsy, anaphylaxis, severe anemia, viral meningitis, severe back pain, hematuria, loss of strength, and sensitivity disorder. The most frequently occurring AEFIs after biHPV vaccination are related to convulsions and neuralgia and were noticeably lower than those of the qHPV vaccine66.
Go to :
IV. Future of HPV Prophylactic Oral Cancer Vaccines
More recently, many researchers have focused on strategic ways to increase protection against HPV infection. Although several subtypes of HPV-positive oral cancer have better prognosis that HPV-negative cancer, fundamental research trends support a prophylactic vaccination strategy for HPV-originating pathologic conditions. Many companies have shared recent research on HPV prophylactic vaccines. A nine-valent HPV vaccine67 has showed equivalent protection against the four types in the qHPV vaccine and greater efficacy against five additional subtypes of cervical cancer, namely, HPV 31, 33, 45, 52, and 58. If cost-effective, this vaccine could extend the spectrum of protection against cervical and oral cancer available through vaccination6768. Other cost-effective investigations have indicated that L1 VLPs can be generated in alternative vectors such as the yeast Pichia pastoris or in plants69. Improvements in vaccine storage and easier administration methods through nasal sprays or patches could also result in easier and cheaper vaccine accessibility70.
1. Characteristic consideration for oral cancer vaccination
The immune system and similarities between the oral and cervical mucosa should be considered when evaluating the characteristics of HPV oral cavity infection. During HPV infection, local or systemic viral antigen presentation to the immune system by antigen-presenting cells is minimal, allowing infection to persist for months or even years without clinical recognition71. It is well known that immune responses play an important role in HPV infections; skin warts often regress spontaneously, and a higher incidence of skin or mucosal infections induced by HPV is found in immunodeficient subjects72. In immunocompetent hosts, immune responses to HPV-related diseases are generally weaker compared to those seen in other viral infections, presumably because the HPV replication cycle takes place inside mature keratinocytes, which continuously remove mature virions. As a result, HPV tends to cause cell proliferation rather than cell lysis73.
The developmental mechanism of oral HPV infection may start in the oral mucosa, which is histologically similar to the uterine cervix, other lower genital tract mucosa, and skin. HPV invasion and transformation of the oral epithelium to multiple lesions are attributed to the histology of the oral cavity, which is lined by a mucous membrane consisting of a stratified squamous epithelium and lamina propria made up of dense connective tissue. The lining of the gingiva, hard palate, and dorsum of the tongue are completely keratinized with a superficial horny layer, whereas the epithelia of the lip, cheek, vestibular fornix, alveolar mucosa, floor of the mouth, and soft palate are non-keratinized. Palatal mucosa can be divided into three distinct anatomical and physiologic zones: the fibro-mucosa of the palatal shelves, lining the palatal vault in the midline; the maxillary fibro-mucosa, lining the area between the midline palatal mucosa and the gingival; and the gingival fibro-mucosa, lying laterally between the maxillary mucosa and the teeth. Based on these morphological similarities, the presence of both mucosal and cutaneous HPV types could be anticipated in different squamous cell lesions of the oral mucosa. Such benign oral lesions include squamous cell papilloma, condyloma, and focal epithelial hyperplasia (FEH), which are linked with HPV27.
Once inside the epithelial basal layer, HPV replicates in the nuclei of infected cells, leading to maturation of virions in the suprabasal epithelial cell layers7. HPV infection is highly transmissible, with a variable incubation period that can culminate in latent infection with low HPV DNA copy-number in basal cells, insufficient to support transmissibility in active subclinical infection without clinical signs, or in clinical infection leading to benign, potentially malignant, or malignant epithelial lesions3. The epithelial areas of the upper aerodigestive tract display the greatest susceptibility to HPV due to the easy exposure of basal cells to HPV infection. The presence of HPV in the oral mucosa suggests that, as in cervical cancer, HPV infection plays a similar role in the transformation of the oral epithelium, targeting the p53 and pRb tumor suppressor pathways and resulting in cell cycle alteration. HPVs have a strong affinity for squamous epithelial cells and are associated with a wide range of proliferative epithelial lesions 1011. Persistent HPV infection in the oral mucosa might increase the risk of oral cancer in a population younger than that typically affected by HPV-independent oral cancer. The use of chewable tobacco products, such as quid and betel nut, also increase the risk of oral cancer.
Considering these factors, benign hyperplasia lesions with low malignant potential in the oral mucosa of healthy adults may not proceed to malignancy without HPV infection. Prophylactic vaccinations act primarily by inactivating HPV before stimulating humoral immunity through host cell infection. Therefore, it is important to determine the appropriate therapeutic and prophylactic vaccination approaches to effectively protect against many of the HPV subtypes.
Based on our review, if the anatomical site is not in the tonsil or base of the tongue, HPV vaccination is not necessary to prevent oral cancer. The majority of the intraoral mucosa already contacts surrounding oral structures, including the tongue, teeth, prostheses, buccal mucosa, and dentures, so HPV penetration to the healthy oral mucosa is not possible without microscopic mucosal lesions. Even before HPV vaccination, the first line of defense in the prevention of oral cancer is maintaining a normal and healthy oral cavity with clean oral hygiene and without precancerous lesions. To meet the therapeutic goals of HPV vaccines, vaccination should evoke cell-mediated immunity and prevent the progression of precancerous lesions or induce the regression of existing lesions. Unfortunately, these therapeutic goals were not effective, and these approaches could be regarded as chemotherapy and immune therapy. Because oral epithelial lesions can result from HPV infection, even in immunocompetent healthy people with clear oral mucosa, previous vaccination is beneficial for preventing HPV-related lesions such as warts, papilloma, or FEH. Oral cancer can develop through different mechanisms than cancer of the tonsil or base of the tongue.
To date, many clinicians and researchers include oral cancer in the term OPC or HNSCC. In addition, oral cancer has a somewhat confusing definition that includes salivary gland cancer and oral cavity-originating sarcoma. Oral cancer terminology must be clarified if we are to evaluate HPV infections and the efficacy of HPV vaccination in the oral cavity. To develop oral cancer vaccination from VLPs of HPV subtypes, preventive goals should focus on avoiding HPV-related lesions. The current HPV vaccination strategy for cervical cancer will prevent development of some oral squamous cell cancer, as broadly defined, as well as some anogenital carcinoma, such as anal, penile, and vulvar cancers.
2. Effectiveness of oral cancer vaccination
HPV infection by HPV 16 and 18 accounts for about 70% of cervical cancer cases worldwide, with relatively small region-specific differences. More than 90% of HPV-associated non-cervical cancers are also attributable to HPV 16 and 18, with HPV 16 accounting for the vast majority of cases456729. Smaller trials of the qHPV vaccine in males have demonstrated protection against genital warts and premalignant anal neoplasia74. Both in Europe and the USA, the rising incidence of OPSCC, including tonsillar and tongue base cancers, has been attributed to an epidemic of HPV infection75. A decrease in the incidence of cancer at HPV-unrelated sites has been reported in Japan, where about 35% of OPC and 25% of other oral cancers are HPV-positive76. The possibility of vaccination against HPV-positive OSCC, including tonsil- and tongue-based cancers, was reported to be effective for decreasing the prevalence of oral HPV infection in middle-aged adults even though vaccine efficacy against oral HPV infections is presently unknown75.
Extension of vaccination to men, particularly men who have sex with men, could further reduce the population prevalence of HPV and provide direct protection to men against genital warts and anal, penile, and OPC. Although the relationship between HPV infection and oral cancer has been well documented, oral cancer has also been linked to different behaviors such as smoking, excessive alcohol consumption, poor oral hygiene, irritation of ill-fitting dentures or prostheses, poor nutrition, and chronic oral infections by microorganisms. Chewing betel, paan, and areca are also known risk factors of oral cancer development. The Karachi South region in Pakistan has the second highest incidence of oral cancer (greater than 17%) according to World Health Organization statistics, due to heavy use of betel, paan, Areca, and Gutka. Oral cavity exposure to these chewable tobaccos causes abrasions and a weak mucosal surface, increasing susceptibility to HPV infections and consequent precancerous and cancerous lesions77.
Several oncogenes are activated as a result of DNA mutation, which can occur from the processes described above, regardless of HPV infection. HPV-infected virions can penetrate through the weakened mucosae of the uterine cervix, lower genitalia, and oral cavity, and non-healing oral cavity ulcerations are susceptible to perforation by these virions. Regardless of HPV infection, it is important to prevent mucosal weakness to avoid carcinogenesis. If ulcerations that do not heal well, including OLP, OL, OE, and nicotine stomatitis, are diagnosed and cured in the earliest stages, such initial treatment could be effective at preventing cancer without HPV vaccination.
Go to :
V. Conclusion
Several suggestive and consistent correlations between HPV infection and oral cancer have been reported worldwide. However, many factors such as controlled study groups, investigation methods, racial and sex differences, and specific anatomical sites of the oral cavity must be considered and classified in order to avoid any independent results. Maxillo-facial surgeons that are responsible for managing patient survival following oral cancer should be knowledge about HPV vaccination and informed of developments in technology for reducing HPV-related infections and cancers.
Go to :
 XML Download
XML Download