

 PDF
PDF ePub
ePub Citation
Citation Print
Print


Long QT syndrome (LQTS) is an inherited cardiac disease characterized by a prolonged heart rate-corrected QT (QTc) interval on the electrocardiogram (ECG). Besides genetic factors, several other conditions can lengthen the QTc interval such as electrolyte imbalance, use of QTc-prolonging drugs, and structural heart diseases.
To date, 15 genes (KCNQ1, KCNH2, SCN5A, ANK2, KCNE1, KCNE2, KCNJ2, CACNA1C, CAV3, SCN4B, AKAP9, SNTA1, KCNJ5, CALM1, and CALM2) have been implicated in LQTS. KCNQ1, KCNH2, and SCN5A are known to be responsible for 60–75% of genotype-positive LQTS cases [12], while the other 12 genes linked to LQTS susceptibility collectively account for <5%. Because genetic predisposition often factors in cases of acquired LQTS, genetic profiling is important, even for patients with mild QTc interval prolongation [3].
Here, we applied targeted sequencing based on a multigene panel containing known LQTS-associated genes to investigate the genetic background of patients with prolonged QTc interval, but negative for mutations in KCNQ1, KCNH2, and SCN5A by Sanger sequencing. The multigene panel also included 67 genes related to other cardiac diseases, which were used to target patients without pathogenic variants of the LQTS-associated genes. For this study, 30 participants were selected among patients with prolonged QTc interval who underwent genetic testing at the Seoul National University Hospital, Korea, between 2005 and 2011. DNA samples had been archived after the routing screening of KCNQ1, KCNH2, and SCN5A variants for the purpose of this research. Most participants exhibited prolonged QTc interval on the resting ECG (>450 ms) and/or positive results on the provocative tests; a few patients with borderline QTc intervals, but suspected LQTS, were also included. The study was approved by the institutional review board of Seoul National University Hospital.
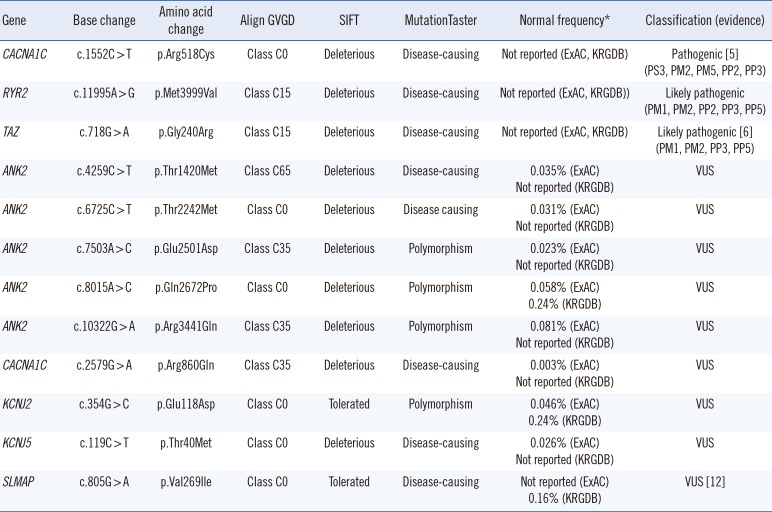
The multigene test panel included 13 known LQTS-associated genes (AKAP9, ANK2, CACNA1C, CAV3, KCNE1, KCNE2, KCNH2, KCNJ2, KCNJ5, KCNQ1, SCN4B, SCN5A, and SNTA1) and 67 genes related to other cardiac diseases (see Supplemental Table S1). DNA samples were enriched using the TruSeq Custom Enrichment Kit and sequenced with MiSeq (Illumina, Inc., San Diego, CA, USA); sequencing data were analyzed using the NextGENe® Software (Softgenetics, State College, PA, USA). Pathogenic/likely pathogenic variants or variants of uncertain significance were further confirmed by Sanger sequencing. Copy number variation was not analyzed. Exonic variants with non-synonymous changes and intronic variants in 10-bp exon-flanking regions were analyzed. Each variant was assessed by considering allele frequencies in normal controls from the 1,000 Genomes database, Exome Aggregation Consortium (ExAC), and Korean Reference Genome DB (KRGDB) and in silico prediction results (Align GVGD, SIFT, and MutationTaster). The highest minor allele frequency (MAF) reported in the databases for any population was taken into account, and variants with MAF >0.1% were filtered out. Each retained variant was classified according to the American College of Medical Genetics (ACMG) guidelines [4]. For previously reported variants, segregation and functional test results were reviewed. When no pathogenic or likely pathogenic variants were found among the LQTS-linked genes, the genes related to other cardiac diseases were sequentially analyzed.
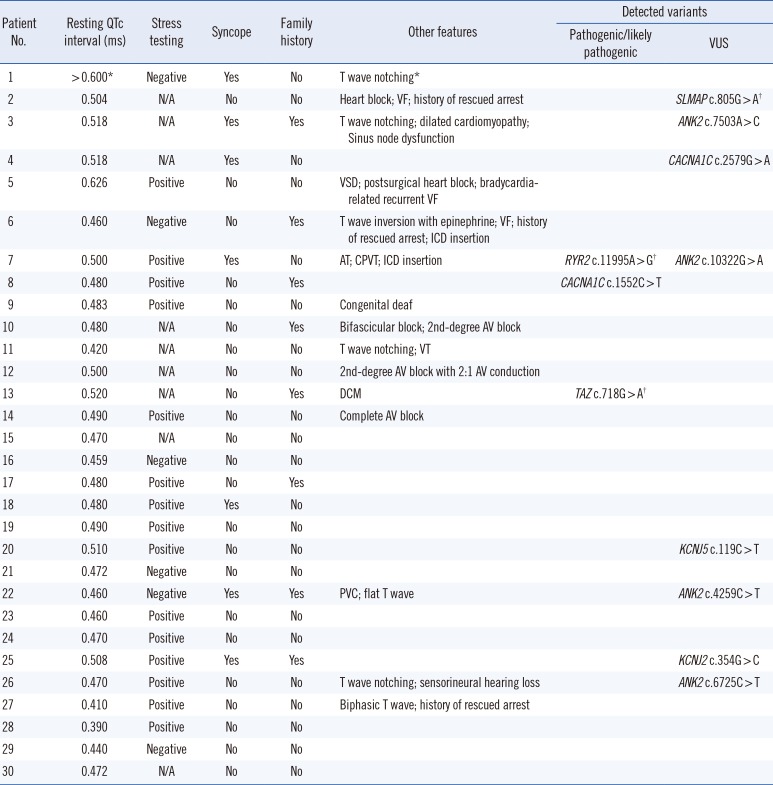
The average coverage depth in target regions of the multigene panel was 235×, representing 1,831 exons in total; 99.77% and 99.95 % of the bases had ≥30× and ≥5× coverage, respectively, the latter was the minimal level of acceptable coverage considered in this study. Regions with coverage <5× were not subjected to Sanger sequencing. The median patient age at examination was 10 years (1 month–30 years), and the average QTc interval was 485 ms (468–502 ms; 95% confidence interval). Of the study participants, only one (patient No. 8) was confirmed to have a pathogenic variant of an LQTS-related gene, CACNA1C c.1552C>T (p.Arg518Cys), which was reported to be associated with Timothy syndrome (Table 1) [5]. This patient showed a phenotype similar to that of an originally reported proband: he had a ventricular septal defect (VSD) detected at birth and QTc interval prolongation of 480 ms accompanied by notched T-wave detected on his ECG at age five. The patient did not have any other extracardiac symptoms frequently found in Timothy syndrome, which could have been caused by the pathogenic variant in the CACNA1C gene. He had a family history of sudden cardiac death, but none of the other family members were tested (Table 2).
Next, genes related to other cardiac diseases were examined in the rest of the patients. One patient (No. 7), who had baseline QTc interval prolongation of 500 ms, exhibited polymorphic ventricular tachyarrhythmia, suggesting a diagnosis of catecholaminergic polymorphic ventricular tachycardia (CPVT). He was found to carry the ANK2 c.10322G>A (p.Arg3441Gln) variant of uncertain significance; however, an examination of the CPVT-related genes revealed that he also harbored a likely pathogenic variant, RYR2 c.11995A>G, (p.Met3999Val), which could have caused CPVT.
Patient No. 13, who developed dilated cardiomyopathy with QTc interval prolongation of 520 ms, carried a likely pathogenic variant, TAZ c.718G>A (p.Gly240Arg), previously reported as a causative mutation in infantile dilated cardiomyopathy [6]. The patient's deceased brother had been similarly diagnosed.
Prior to this study, we tested 57 patients with prolonged QTc interval for pathogenic point mutations and large deletions/duplications in KCNQ1, KCNH2, and SCN5A. Twenty-six patients (45.6%) were identified with pathogenic variants associated with QTc interval prolongation: 14 carried pathogenic mutations in KCNQ1 (24.6%), six in KCNH2 (10.5%), and six in KCNH2 (10.5%) (unpublished data). The detected proportion of pathogenic variants (45.6%) was relatively low compared with that in a previous study demonstrating that pathogenic variants in KCNQ1, KCNH2, and SCN5A accounted for nearly 75% of clinically defined LQTS cases and up to 80%, if copy number variant or genomic rearrangement data were included [7]. Another study tested a panel of 12 LQTS-related genes and reported a molecular diagnostic level of 30%, which was probably due to the inclusion of patients with borderline QTc intervals without other clinical symptoms [8]. More stringent criteria considering longer QTc intervals would increase the detection rate; however, in clinical laboratory settings, physicians may request diagnostic testing for patients with prolonged QTc interval and an intermediate probability of LQTS. Therefore, mutation analysis of genes related to a broader spectrum of cardiac diseases that can cause QTc interval prolongation should improve the diagnostic rate.
In this study, we detected pathogenic or likely pathogenic variants relevant to QTc interval prolongation in three out of 30 patients (10%) negative for KCNQ1, KCNH2, and SCN5A pathogenic variants. Only one patient was confirmed to have a pathogenic variant in an LQTS-related gene. When we expanded the targets to other cardiac disease-related genes, we detected likely pathogenic variants in two patients who showed cardiac manifestations other than prolonged QTc interval. Similar to the patient No. 7, who carried RYR2 c.11995A>G, several other cases with RYR2 mutations suspected of LQTS were diagnosed as CPVT in a previous study [9]. It should be noted that RYR2 has been proposed as a candidate gene involved in LQTS pathogenesis as it exhibits interactions with several genes with an established role in LQTS pathogenesis [1011].
In addition, the SLMAP c.805G>A variant, originally reported in a Japanese patient with Brugada syndrome [12], was detected in one of our participants who exhibited an ECG with a complete AV block, but did not exhibit Brugada syndrome features. A previous functional analysis study showed that SLMAP c.805G>A affects the membrane expression of cardiac sodium channel hNav1.5 and reduces hNav1.5-dependent current [12]. However, we found that this variant had a MAF of 0.16% by KRGDB, which was higher than expected for the disorder; therefore, it was recategorized as a variant of uncertain significance.
Because multigene panel sequencing has become widely available, mutation screening of cardiac disease-related genes that may cause QTc interval prolongation can be conducted in parallel, providing more comprehensive molecular analysis and improving the diagnosis rate in patients negative for mutations in LQTS-related genes. Incidental findings of well-described genes included in the expanded gene panel should be taken into consideration, especially if they are present in the minimum list recommended by the ACMG [13].
In conclusion, the multigene panel sequencing performed in this study enabled comprehensive screening of genetic variants with possible involvement in prolonged QTc interval and helped identify additional patients with genotypes that may lead to QTc interval prolongation. Thus, this approach expands the spectrum of genetic causes underlying QTc interval prolongation and can help prevent adverse outcomes, such as sudden cardiac death, in patients and their relatives [14].
 XML Download
XML Download