

 PDF
PDF ePub
ePub Citation
Citation Print
Print


INTRODUCTION
BCR/ABL1-negative myeloproliferative neoplasms (MPNs) include polycythemia vera (PV), essential thrombocythemia (ET), prefibrotic primary myelofibrosis (prePMF), and overt primary myelofibrosis (PMF) [12]. They are among the most frequent hematologic neoplasms, usually affecting the elderly population. Thrombosis, hemorrhage, evolution to myelofibrosis, and leukemic transformation represent the clinically relevant issues in the course of disease [3]. Especially in PMF, life expectancy was markedly lower than that in an age and sex-matched population in many studies [45].
The 2016 WHO classification system divided PMF into prePMF and overt PMF on the basis of the absence or presence of grade ≥2 bone marrow fibrosis [1].
The most commonly recognized mutation in BCR/ABL1-negative MPN is JAK2 V617F, followed by the CALR and MPL mutations [67]. There has been continued debate on how to subtype BCR/ABL1-negative MPNs since none of the three major mutations are specific to any single subtype of MPN. Therefore, with this incomplete understanding of the molecular features of each subtype, an accurate characterization and standardization of morphological features is still being emphasized as a key to differentiate subtypes as per the 2016 WHO classification [1]. Furthermore, the disease subtypes often show considerable overlap in clinical and histopathological characteristics, especially in the early course of the disease. The differentiation between ET and prePMF has been particularly challenging, and diagnostic discrepancies exist even among experts [89]. The peripheral blood and bone marrow findings in patients with early or cellular phase PMF (prePMF) are similar to those of patients with ET. Hence, it is difficult yet imperative to discriminate between ET and prePMF as the clinical outcome in patients with prePMF is considerably worse than in those with ET [1011121314].
Even though the 2016 WHO classification has provided criteria for the diagnosis of prePMF [1], bone marrow pathology provides major clues for the differential diagnosis of prePMF from ET. For both ET and prePMF, the histological hallmarks lie in the megakaryocyte (MK) population: ET is marked by proliferation of enlarged and mature MKs with deeply lobed and hypersegmented nuclei, whereas prePMF is characterized by proliferation of markedly atypical MKs that often form clusters. Megakaryocytic atypia involves maturation defects along with atypical nuclear features such as hypolobulation, hyperchromicity, irregular folding, and dense clustering [15]. However, the morphological evaluation of bone marrow specimens is subjective, and the reproducibility has been questioned [89].
Therefore, we aimed to determine whether an immunohistochemical assay would allow for a more objective histopathological assessment of bone marrow MKs. Since each subtype of BCR/ABL1-negative MPN is characterized by well-defined features during MK differentiation, we searched for immunohistochemical markers that could highlight abnormalities in MKs. GATA1, the founding member of the GATA family, is a DNA- and protein-binding transcription factor that is essential for erythroid precursor differentiation and MK maturation [1617181920]. Precursor cells of MK lineage lacking GATA1 may arrest in maturation and undergo unrestrained proliferation [1617181920]. Some studies reported that defective expression of GATA1 in MKs may be associated with myelofibrosis [1617181920]. Moreover, the development of myelofibrosis has been documented in mice with reduced GATA1 gene expression [1719]. We investigated GATA1 expression at diagnosis and during follow-up for better characterization of each subgroup of BCR/ABL1-negative MPNs.
METHODS
1. Study and control groups
To select study subjects, we re-evaluated bone marrow slides and reviewed clinical and laboratory data of patients with BCR/ABL1-negative MPN, MPN, unclassifiable (MPN-U), or myelodysplastic syndrome/MPN, unclassifiable (MDS/MPN-U) between 1998 and 2015 at Ewha Womans University Mokdong Hospital, Seoul, Korea. On application of the 2016 WHO classification [1] after a thorough re-evaluation by two pathologists, 14 patients were reclassified from their original diagnoses, which were based on the 2001 or 2008 WHO classification [2122]. Five patients with ET, two with MPN-U, and three with MDS/MPN-U were reclassified as prePMF, and three MPN-U and one MDS/MPN-U were reclassified as PV.
Forty-nine patients who had BCR/ABL1-negative MPN (nine PV, 17 ET, 12 prePMF, and 11 PMF) according to the 2016 WHO classification were included in this study. Among 17 patients with ET, four had megakaryocytic atypia favoring the diagnosis of prePMF. In one of these four patients, MF-1 fibrosis was present, which was rare in ET. However, these four patients were finally diagnosed as having ET because the diagnostic criteria for prePMF were not met. This study was exempted from review by the Institutional Review Board of Ewha Womans University, and all information was de-identified.
The control group for GATA1 stain comprised 40 patients, of which nine had BCR/ABL1-positive chronic myeloid leukemia (CML), 16 had lymphoma without bone marrow involvement, and 15 had benign hematologic diseases (BHD) (eight idiopathic thrombocytopenic purpura, two iron deficiency anemia, and five normocellular marrow).
Electronic medical records were reviewed for retrospective collection of relevant clinical and laboratory data including the results of cytogenetics and JAK2 mutation. The JAK2 V617F mutation was identified in all patients with PV, 53% of those with ET, 67% of those with prePMF, and 40% of those with PMF. Leukemic transformation was observed in 8% patients with prePMF, whereas no patients with ET developed acute leukemia. Moreover, 67% patients with prePMF eventually developed fibrosis of MF>2 in the course of the disease over 19 to 82 months (median 55.5 months), whereas no patient with ET developed overt fibrosis. The baseline characteristics of study group are summarized in Supplemental Data Table S1.
The follow-up period varied from nine to 167 months (median 86.5 months). No patient received JAK2 inhibitor therapy or stem cell transplantation during the follow-up period.
2. Cytogenetics and molecular assays
Cytogenetic analysis was performed by unstimulated 24- and/or 48-hour culture and conventional G-banding techniques using the fresh bone marrow aspirates obtained at diagnosis. For molecular assays, genomic DNA was extracted from fresh bone marrow aspirates or stored bone marrow cell pellets fixed for cytogenetic analysis using a DNA purification kit (Qiagen, Hilden, Germany). An allele-specific, real-time PCR assay (Real-Q JAK2 V617F kit, BioSewoom, Seoul, Korea) was used for qualitative analysis of the JAK2 V617F mutation.
Quantification of JAK2 V617F allele burden was performed in 25 patients with JAK2 mutation. Fifteen patients without JAK2 mutation were subjected to molecular analysis of CALR and MPL mutations.
The allele burden of JAK2 V617F was measured as the percentage of mutant allele with respect to total JAK2 (mutant plus wild-type), using the JAK2 V617F quantification kit (BioSewoom). The CALR mutations were assessed by Sanger sequencing. Exon 9 of CALR was amplified using the following primers: forward, 5′-CAT TCA TCC TCC AGG TCA AG-3′ and reverse, 5′-TGA AAG TTC TCG AGT CTC ACA G-3′. PCR was performed with the following cycle profile: an initial 5 minutes denaturation at 94℃, 35 cycles of 94℃ for 30 seconds, 60℃ for 30 seconds, 72℃ for 30 seconds, and a final 7 minutes extension at 72℃.
The MPL W515L/K mutations were assessed by real-time PCR (Real-Q MPL W515L/K screening kit, BioSewoom), according to the manufacturer's instructions.
3. Immunohistochemical assays of GATA1 expression
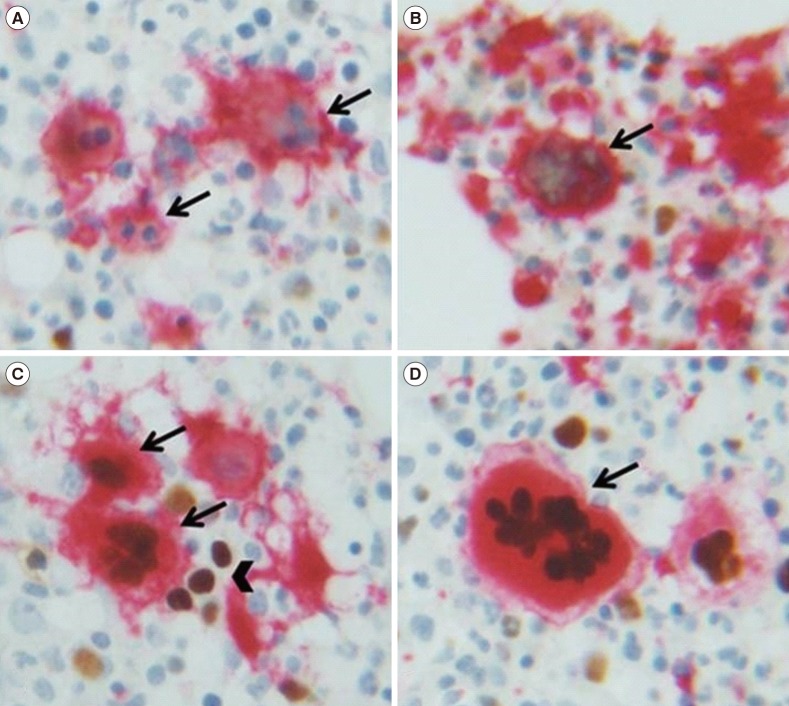
The archived formalin-fixed and paraffin-embedded bone marrow biopsies were used for GATA1 immunohistochemical stain. Bone marrow biopsy samples obtained at diagnosis from 49 patients with BCR/ABL1-negative MPN were stained and analyzed, and serial analyses were available for 18 patients. To better identify MKs, dual staining with GATA1 and CD61 was conducted. The immunohistochemical staining of GATA1 was performed using GATA1 monoclonal antibodies (GATA1 [D52H6] XP rabbit monoclonal antibodies, Cell Signaling Technology, Danvers, MA, USA), and the cytoplasm of MKs were stained with CD61 mouse monoclonal antibodies (EMD Millipore Co., Temecula, CA, USA). Antibodies were used at 1:100 dilutions. All MKs (median, 72; range, 10–345 MKs per slide for one patient) were evaluated at 400× magnification. Bone marrow specimens with less than 10 MKs were excluded from the GATA1 immunohistochemical assay analysis. MKs were classified as GATA1-positive, if the nuclei were clearly GATA1-positive (brown) with CD61-staining (red) in the cytoplasm.
GATA1 expression was semi-quantitatively evaluated in terms of GATA1 score, which was obtained by multiplying the intensity score by the positive cell percentage score [2324]. GATA1 immunostaining intensity was rated on a scale from 0 to 3: negative, 0; weak, 1; moderate, 2; and strong, 3 (Fig. 1). The percentage of positive MKs (the number of GATA1-stained MKs×100/total number of MKs) was graded from 0 to 4: less than 1%, 0; 1–20%, 1; 21–50%, 2; 50–80%, 3; and 81–100%, 4.
4. Statistical analysis
The differences in continuous variables (GATA1 score, age, hemoglobin level, white blood cell count, platelet count, lactate dehydrogenase, and JAK2 V617F allele burden) between more than three groups were evaluated using the Kruskal-Wallis test and subsequently calculated with post hoc Mann-Whitney test between each two groups. When the independent variables were fewer than three, the Mann-Whitney test was used. Categorical variables (sex; presence of leukoerythroblastosis, splenomegaly, myelofibrosis, cytogenetic abnormalities, or any specific mutation; progression to acute leukemia; and development of fibrosis) were compared between groups using chi-square tests, and subsequent P values were calculated using Fisher's exact test.
All statistical analyses were performed with MedCalc for Windows, version 16.4.3 (MedCalc Software, Ostend, Belgium). P values <0.05 were considered statistically significant.
RESULTS
1. Molecular analyses
The CALR mutation was detected in 18% patients with ET, 22% patients with prePMF, and 30% patients with PMF. Furthermore, 29% patients with ET, 11% patients with prePMF, and 30% patients with PMF were negative for all three mutations (JAK2, CALR, and MPL). There were no differences in the frequency of CALR or triple negative mutations among ET, prePMF, and PMF.
The median value and the range of the allele burden of JAK2 V617F at initial diagnosis for each subtype of BCR/ABL1-negative MPN are presented in Supplemental Data Table S1. The allele burden of JAK2 V617F differed significantly between the groups (P=0.022), and subsequent post hoc comparisons verified that it was lower in ET than in PV (P<0.001), prePMF (P=0.022), or PMF (P=0.033). No significant difference in the JAK2 V617F allele burden was observed between the disease groups other than ET.
JAK2 V617F allele burdens of more than 50% were observed in 89% patients with PV, 50% patients with prePMF, and 67% patients with PMF, but none was observed in patients with ET.
2. GATA1 expression analysis
1) Comparison of GATA1 expression according to disease group
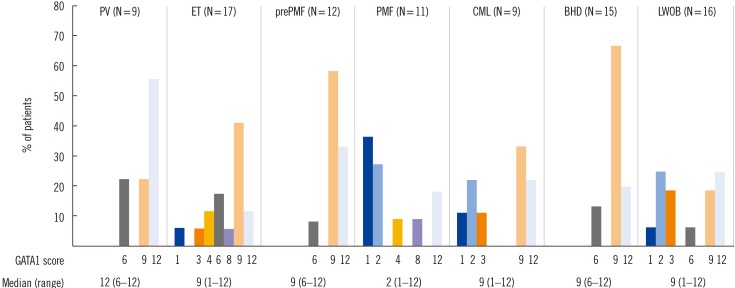
The GATA1 score differed significantly between disease groups (P=0.003). The cut-off score for normal GATA1 expression was set at 6 based on the finding that no patient in the BHD group showed a score less than 6. A GATA1 score less than 6 was considered as low GATA1 expression.
PV and prePMF showed a GATA1 score distribution similar to that of BHD. For PMF, the GATA1 score was significantly lower than that of PV, prePMF, or BHD. Further, 73% patients with PMF scored less than 6, whereas no patients had a GATA1 score less than 6 in the PV or prePMF groups (Fig. 2).
Patients with ET showed a heterogeneous GATA1 score distribution. Features that favored the diagnosis of prePMF, such as increased lactate dehydrogenase and/or megakaryocytic atypia, were present in four patients with ET who scored less than 6 at diagnosis. MF-1 fibrosis was present in one of these four patients, which was rare in ET. However, they were classified as ET because the patients either had normocellular marrow or the minor criterion for prePMF was not met. No statistically significant difference in GATA1 score was found between the ET and prePMF groups. GATA1 expression in the CML and lymphoma without bone marrow involvement groups was heterogeneous, as shown in Fig. 2.
2) Comparison of GATA1 expression with respect to myelofibrosis grade, mutation profile, or JAK2 V617F allele burden
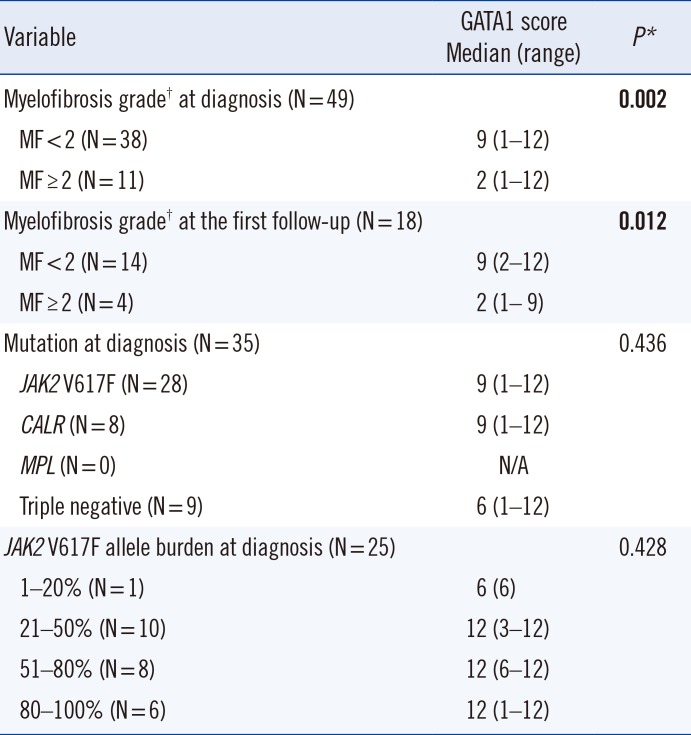
GATA1 expression was compared between two groups in the BCR/ABL1-negative MPNs with specimens obtained at diagnosis: one with WHO myelofibrosis grade of MF≥2 (MF–2 or 3) and the other with MF<2 (MF–0 or 1). The median value of GATA1 score in the MF≥2 group was significantly lower than in the MF<2 group (P=0.002). GATA1 expression according to myelofibrosis grade (MF≥2 vs MF<2) at the first follow-up yielded results similar to those obtained at diagnosis (P=0.012) (Table 1).
When GATA1 scores were compared according to mutation profile (JAK2, CALR, and triple negative mutations) at diagnosis, the difference between groups was not significant (P=0.436), although the median value of the GATA1 score was lowest in the triple negative group (Table 1).
The median value of the GATA1 score tended to increase as JAK2 V617F allele burden increased, but no statistically significant difference was observed (P=0.428) (Table 1).
In the PV, ET, and prePMF groups, no patient had both JAK2 V617F burden>50% and GATA1 score<6 at diagnosis, whereas 33% patients with PMF showed both traits, as shown in Supplemental Data Table S2.
3) Serial analysis of GATA1 expression and clinical course
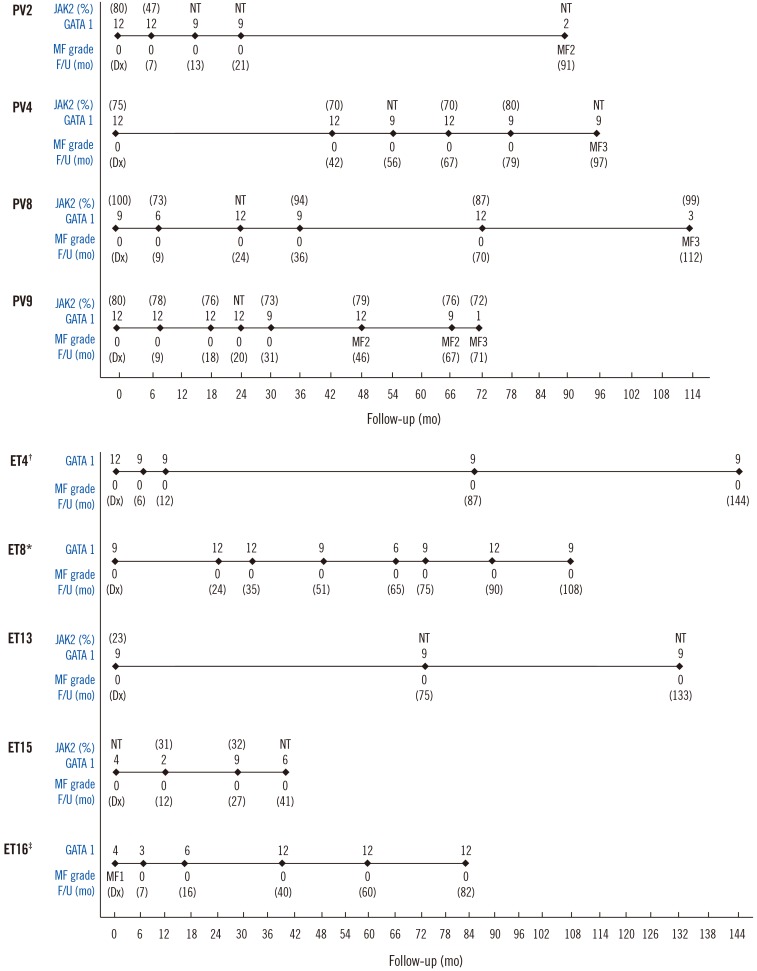
Figs. 3 and 4 depict GATA1 expression and clinical events in patients with follow-up bone marrow biopsies. In patients with PV, the GATA1 score on diagnosis was 9 or 12 and remained higher than 6 throughout the disease course until the development of overt myelofibrosis (MF≥2). GATA1 expression decreased to scores of 1–3 with progression of myelofibrosis, except for one patient (patient PV-4).
Among the five patients with ET, three (patients ET-4, ET-8, and ET-13) showed a steadily high GATA1 score (6 or higher). Two patients (ET-15 and ET-16) showed low GATA1 expression at diagnosis with a score of 4, but the scores increased to 6 or higher in the follow-up course. Patient ET-16 initially showed MF-1 myelofibrosis and low GATA1 score, but the fibrosis was reversed during the follow-up course along with increase in GATA1 score.
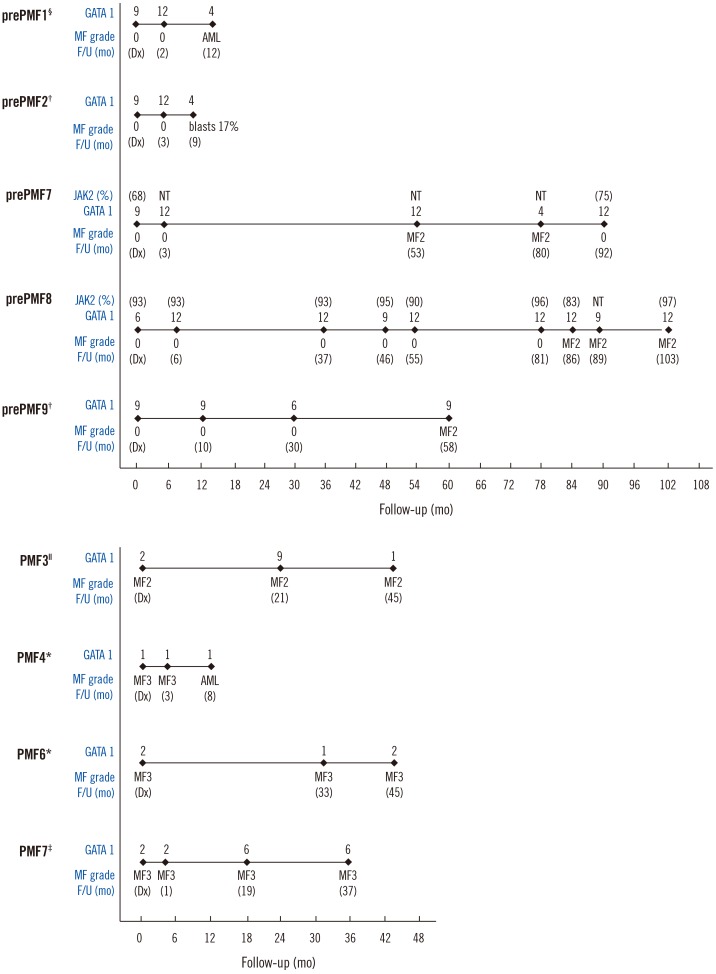
The GATA1 score decreased from 9 to 4 when the disease transformed to acute myeloid leukemia for one patient (prePMF-1). The GATA1 score of another patient (prePMF-2) decreased from 9 to 4 and the blasts increased in number (17% in bone marrow differential counts). However, in three patients with prePMF (prePMF-7, prePMF-8, and prePMF-9), the GATA1 scores remained high (score of 9–12) even when the myelofibrosis developed to MF-2.
In patients with PMF (PMF-4 and PMF-6), the GATA1 score was 1 or 2 at diagnosis, which remained unchanged during follow-up with leukemic transformation or extensive fibrosis. The GATA1 score of two patients (PMF-3 and PMF-7) was 2 at diagnosis, which increased during follow-up without improvement of fibrosis on bone marrow biopsy.
JAK2 V617F allele burdens did not show significant changes during follow-up in patients with PV, ET, or prePMF although GATA1 expression changed with development of myelofibrosis or leukemic transformation.
DISCUSSION
We demonstrated that GATA1 expression was significantly lower in PMF than in other subtypes and the allele burden of JAK2 V617F was significantly lower in ET than in other subtypes. GATA1 expression did not differ according to the mutation profiles, but significantly decreased in patients with progressed fibrosis or leukemic transformation.
The frequencies of JAK2 V617F and CALR mutations in PV, ET, and PMF were similar to those found in previous studies [67252627]. Previous studies reported significantly higher allele burdens of JAK2 V617F in PV and PMF than in ET [272829303132333435]. We also demonstrated that the burden of the JAK2 V617F allele in patients with ET was significantly lower than in patients with PV or PMF. The allele burden of JAK2 V617F was significantly higher (P=0.022) in the prePMF group (median, 50.4; range, 31.6–93.1) than in the ET group (median, 29.8; range, 19.7–50.0). A JAK2 V617F allele burden higher than 50% was not detected in any patient with ET, similar to the findings of previous studies [1028293032]. Our results supported that a diagnosis of ET was less likely when a patient's JAK2 V617F allele burden was high (more than 50%) [28293034].
We assessed GATA1 expression in MKs to demonstrate its clinical relevance in distinguishing between BCR/ABL1-negative MPN subtypes, especially prePMF from ET. GATA1 is a transcription factor that regulates maturation and differentiation of erythroid cells and MKs; Owing to genetic alterations, GATA1-low mice are both anemic and thrombocytopenic at birth [1636]. Few studies have shown that the number of MKs with retarded nuclear and cytoplasmic development increased significantly in GATA1-low mice [161736]. Overall, impaired control of both MK proliferation and maturation is conceivably the result of low GATA1 expression. An animal model also showed that mice with defective GATA1 expression eventually developed myelofibrosis [19]. Other studies have also demonstrated decreased GATA1 expression in patients with PMF [1720]. In agreement with these previous studies, we showed that GATA1 expression was reduced in patients with overt myelofibrosis and that more than 60% patients with PMF had low GATA1 expression. GATA1 score less than 6 was observed in 24% (four out of 17) patients with ET, including patients ET-15 and ET-16 with features which favored the diagnosis of prePMF; however, these patients did not fulfill the complete diagnostic criteria for prePMF. We speculated that these patients were in the gray area of diagnosis where it was uncertain whether they truly belonged to the ET or prePMF. Indeed, few studies have shown borderline ET/prePMF in 5–18% patients with ET [3738].
In the serial analysis, GATA1 expression was also low in patients with PV who developed post-PV myelofibrosis during follow-up. The GATA1 score was significantly lower in the BCR/ABL1-negative patients with MPN and myelofibrosis of MF≥2 than in those with MF<2, irrespective of the disease subtype. Two patients with PMF showed unexpected GATA1 score-fluctuation during the follow up course, which warrant further research in future. Interestingly, two patients with prePMF showed progressive decrease in GATA1 expression during leukemic transformation. Since the somatic mutation of GATA1 that reduces GATA1 expression is involved in the accumulation of immature erythroid and MKs progenitor cells [3940], these cells may subsequently acquire additional genetic hits and contribute to multi-step leukemogenesis [3940]. The possible association between reduced GATA1 expression and leukemogenesis should be investigated in the future.
Our study has some limitations. The small patient population for each subtype of BCR/ABL1-negative MPN and the retrospective design of the study did not allow us to draw a definitive conclusion. Since an accurate histological diagnosis is still being emphasized in the 2016 WHO classification, further research on immunohistochemical markers is warranted for better and more standardized morphological evaluation.
In conclusion, although our attempt to accurately distinguish between subgroups using a GATA1 immunohistochemical approach did not achieve statistical significance, our results suggest that GATA1 expression decreases with progressive fibrosis and possibly with leukemic transformation.
 XML Download
XML Download