

 PDF
PDF ePub
ePub Citation
Citation Print
Print


Dear Editor,
Axenfeld-Rieger syndrome (ARS) is a rare autosomal dominant disorder characterized by ophthalmologic anterior segment abnormalities and extraocular findings, including dental anomalies, cardiovascular outflow tract malformations, and craniofacial abnormalities [1]. Mutations in paired like homeodomain 2 (PITX2) and forkhead box C1 (FOXC1) genes, which play important roles in embryonic development, are responsible for causing ARS [2]. Affected individuals with PITX2 mutations typically have systemic abnormalities in addition to ophthalmologic issues, while FOXC1 mutations are mainly associated with ocular lesions [3].
PITX2 is a member of the paired class of homeodomain transcription factors and affects the development of various anterior segment tissues that originate from the neural crest as well as several extraocular tissues such as the heart [4]. Missense, nonsense, and frameshift pathogenic variants of PITX2 have been identified in ARS [5]. These pathogenic variants are thought to affect the expression of PITX2.
In Korea, one family with PITX2-related ARS and two families with FOXC1-related ARS have been reported [678]. Here, we present the clinical findings of a Korean ARS family carrying a novel pathogenic PITX2 variant.
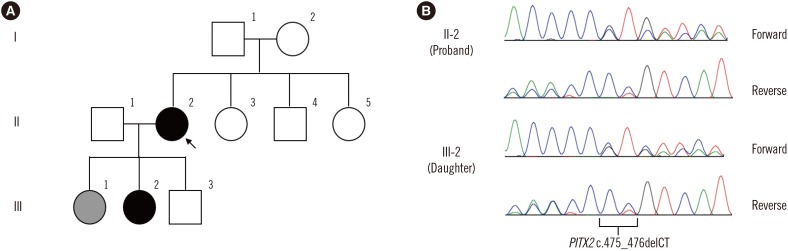
The proband (I-1) was a 46-year-old woman who had been treated for several years for glaucoma in the right eye. The patient had been blind in the left eye for the last 10 years and lacked a detailed medical history. At ophthalmologic examination, the patient showed corneal opacity in both eyes, with more severe presentation in the left eye. Visual acuity was 20/100 in the right eye, with no light perception in the left eye. Intraocular pressure (IOP) was 30 mmHg in the right eye. The right eye showed corneal endothelial decompensation, posterior embryotoxon, iris atrophy, and corectopia. She underwent corneal transplant for the right eye. One year later, she underwent glaucoma surgery because of uncontrolled IOP elevation. The patient had no systemic findings.
The 22-year-old second daughter (III-2) of the proband underwent ophthalmic examination because of her mother's ocular history. Visual acuity was 20/20 in both eyes. Anterior segment examination revealed posterior embryotoxon in both eyes, but no other abnormalities or glaucomatous optic nerve damage were found. Detailed glaucoma investigation, including visual field testing, revealed no abnormal signs. The patient showed microdontia, but no other systemic abnormalities were detected.
Under diagnostic suspicion of ARS, the proband's FOXC1 and PITX2 were sequenced after obtaining informed consent. Genomic DNA was isolated from peripheral blood leukocytes using the Wizard Genomic DNA Purification kit (Promega, Madison, WI, USA). All exons and their flanking intronic regions were amplified by polymerase chain reaction using primers designed by the authors (available on request), and Sanger sequencing was performed using an ABI Prism 3730xl Genetic Analyzer (Applied Biosystems, Foster City, CA, USA). A novel heterozygous 2-bp deletion (c.475_476delCT) in PITX2, which was predicted to result in a frameshift and premature termination of PITX2 (p.Leu159Valfs*39), was detected. No pathogenic FOXC1 variants were detected. Similar genetic analysis of the second daughter confirmed inheritance of the c.475_476delCT (p.Leu159Valfs*39) variant (Fig. 1).
The c.475_476delCT variant was absent in dbSNP (build 149), Exome Aggregation Consortium (http://exac.broadinstitute.org/), and Korean reference genome databases at the time of the study (2017). This PITX2 c.475_476delCT variant can be considered a “pathogenic” variant according to the 2015 American College of Medical Genetics and Genomics and Association guidelines based on the following factors: (1) the variant is expected to cause premature termination (PVS1), (2) the variant is absent in large-population databases (PM2), and (3) patient phenotype is highly specific for the disease (PP4) [9].
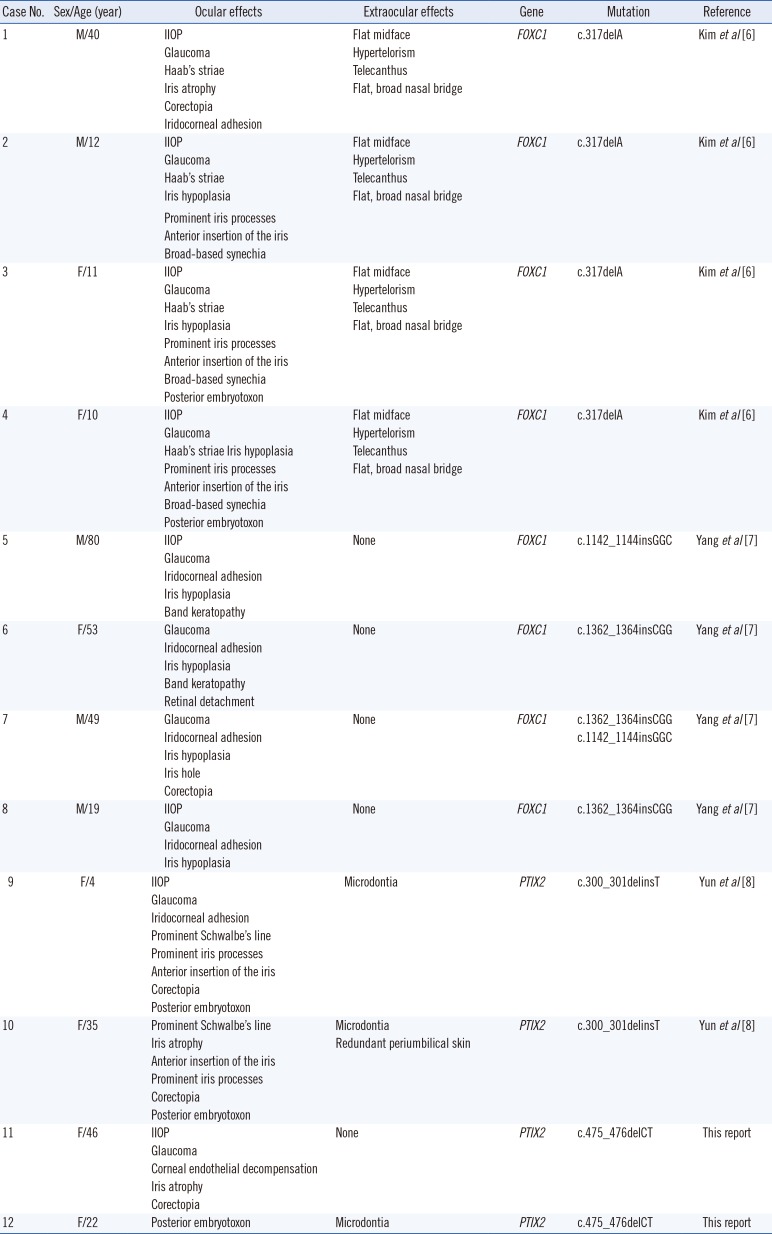
Ten cases of genetically confirmed ARS have been reported in Korea, in addition to the present two cases. The clinical features, including ocular and extraocular symptoms, and molecular features of these cases are described in Table 1.
Glaucoma is observed in approximately 50% of the ARS cases; however, for the proband's daughters (III-1, 2, and 3), no glaucomatous optic nerve damage was detected despite the presence of signs consistent with ARS [10]. The first daughter (III-1) showed microdontia without other systemic abnormalities or ocular signs. Visual acuity and IOP were in the normal range, and anterior segment examination and optic disc were normal. The second daughter (III-2) showed a typical ocular anterior segment finding of ARS, namely, posterior embryotoxon. However, no other ocular findings such as iris atrophy, corectopia, or glaucomatous optic nerve damage were present. Similar to her elder sister, she had microdontia, but no other systemic abnormalities. The third daughter (III-3) underwent ophthalmologic examinations at an outside clinic, but no abnormalities were detected. She had neither microdontia nor systemic abnormalities.
In conclusion, we identified a novel pathogenic PITX2 variant (c.475_476delCT;p.Leu159Valfs*39). This report will deepen our understanding of the genetic background of Korean ARS patients.
 XML Download
XML Download