

 PDF
PDF ePub
ePub Citation
Citation Print
Print


INTRODUCTION
Currarino syndrome (CS; OMIM #176450), first described in 1981, is a rare congenital disorder characterized by the triad of sacral agenesis, anorectal malformation, and the presence of a presacral mass [1]. The most frequent type of sacral agenesis is hemisacrum, which is a pathognomonic sign of CS and is also known as “scimitar sacrum” because of peculiar radiological findings such as sickle-shaped or crescent-shaped sacral deformities. Besides these major signs, developmental delays, neural tube defects, sensorineural deafness, and other features such as depressed nasal bridge, joint hyperextensibility, cataracts, ptosis, microcephaly, and facial dysmorphism have been reported in patients with CS [234]. Phenotypic variability has also been observed in these patients, with some of the major signs not being observed in incomplete cases [56789101112].
The autosomal dominant inheritance pattern of CS was noted by Yates et al [13]. CS is caused by haploinsufficiency of motor neuron and pancreas homeobox 1 (MNX1), located on chromosome 7q36.3 [5141516]. This gene encodes a transcription factor that contains a homeobox domain. Pathogenic variants in MNX1 have been revealed in almost all cases of familial CS, and in approximately 30% of sporadic CS cases [51718]. At least 82 MNX1 heterozygous pathogenic variants have been reported to date [18]; two studies on MNX1 pathogenic variants in Korean patients with CS have been conducted, which included three familial cases and two sporadic cases [919]. However, these pathogenic variants have been identified only in familial cases; moreover, reduced penetrance and variable expressivity of the MNX1 pathogenic variants in CS cases have been reported, even in familial cases [67891011121819]. In this study, we investigated a large number of patients with CS to determine the spectrum of MNX1 pathogenic variants, and described, for the first time, the MNX1 pathogenic variants in Korean patients with sporadic CS.
Go to : 
METHODS
1. Study subjects
In total, 25 index patients were included in this study between July 2014 and June 2015. These Korean patients were diagnosed as having CS and treated at the Department of Pediatric Surgery at Seoul National University Children's Hospital, Seoul, Korea. Medical records were reviewed to collect clinical information. Among these patients, only one patient had a family history of CS, while the remaining 24 patients were classified as sporadic cases of CS upon thorough investigation of family history. This study was approved by the Institutional Review Board of Seoul National University Hospital (No. 1703-125-841). Written informed consent was obtained from all patients and/or their legal representatives.
2. Genetic analysis
Genetic analysis was performed on archival samples. Genomic DNA was extracted from peripheral blood samples using the Gentra Puregene blood kit (Qiagen, Valencia, CA, USA). PCR was performed using newly designed primers specific to the three exons and flanking regions of MNX1. Direct sequencing with in-house primers was performed on an Applied Biosystems 3730xl DNA Analyzer (Applied Biosystems, Foster City, CA, USA) using a BigDye Terminator v3.1 Cycle Sequencing Kit (Applied Biosystems). Sequence variants were identified according to the NM_005515.3 and NP_005506.3 reference sequences using Sequencher 5.0 (Gene Codes Corporation, Ann Arbor, MI, USA). If the result of sequence analysis was negative, gene dosage analysis was performed by using a multiplex ligation-dependent probe amplification (MLPA) kit (SALSA MLPA probemix P277-B2 Human Telomere-10; MRC-Holland, Amsterdam, Netherlands). The MLPA kit contains 14, 11, 11, and 11 probes in the terminal region of chromosomes 5q, 6q, 7q, and 8q, respectively. In silico prediction of missense variants was carried out using Polyphen-2, SIFT, and Mutation Taster [202122]. The splicing effect of variants was predicted using Human Splicing Finder [23]. The variants were classified according to the latest American College of Medical Genetics guidelines [24].
Go to :
RESULTS
1. Clinical features of patients with CS
The study group consisted of 25 index patients (11 males and 14 females). Most patients (21/25) presented the complete CS triad, while the remaining four patients showed only two major CS features without sacral anomalies (Table 1). Patient 1 was the only familial case, and she presented the complete CS phenotype.
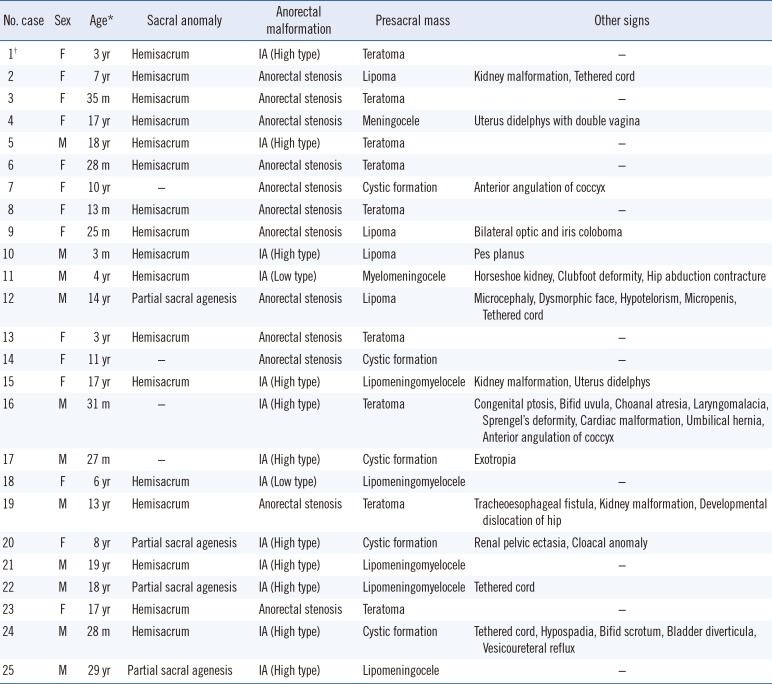
Table 1
Clinical data of patients with Currarino syndrome
![]()
Hemisacrum (17/25) was the predominant sacral anomaly phenotype, followed by partial sacral agenesis (4/25). Anorectal malformations, such as imperforate anus (13/25) and anorectal stenosis (12/25), were observed in all patients. Imperforate anus (9/11) was the most common anorectal malformation in males, while anorectal stenosis (10/14) was predominant in females. Sex-dependent differences were not observed for the other two major CS phenotypes. Among the 13 imperforate anus cases, high type (11/13) was more common than low type (2/13). All patients presented with presacral mass. Teratoma (9/25) was observed predominantly, followed by cystic formation (5/25), lipoma (4/25), lipomeningomyelocele (4/25), and others.
Twelve patients presented with other signs in addition to the Currarino triad. Urogenital malformations, including kidney malformation, uterus didelphys, micropenis, hypospadia, bifid scrotum, bladder diverticula, and vesicoureteral reflux were most commonly observed. Musculoskeletal deformities (Sprengel's deformity, clubfoot, pes planus, and developmental dislocation of the hip), eye complications (coloboma, ptosis, and exotropia), and tethered cord were observed in some cases. Microcephaly and dysmorphic facial features were identified only in one case.
2. Genetic analysis
From MNX1 sequence analysis, we identified seven different pathogenic or likely pathogenic variants, including four frameshift variants (p.Gly98Alafs*124, p.Gly145Alafs*77, p.Gly151Leufs* 67, and p.Ala216Profs*5), two nonsense variants (p.Tyr186* and p.Gln212*), and one missense variant (p.Lys295Gln) (Fig. 1 and Table 2). Except for one missense variant located in exon 3, the other six variants were located in exon 1.
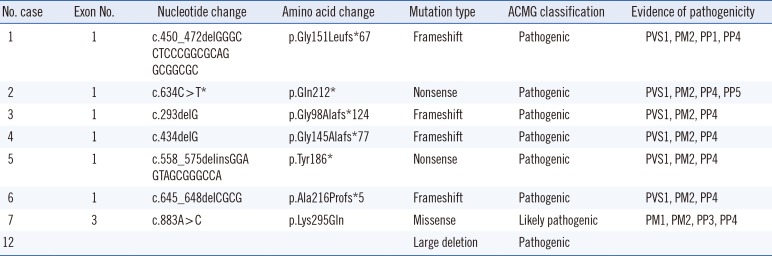
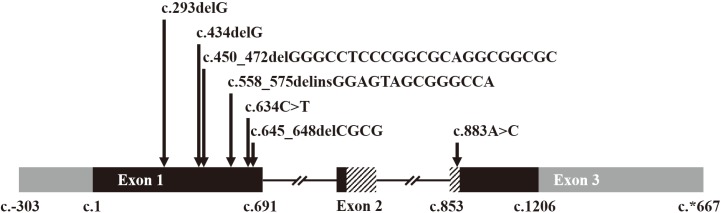 | Fig. 1Distribution of the seven different MNX1 variants. Length of the exons and location of the variants are displayed in proportion to actual length and location, except introns. Black boxes indicate coding regions and gray boxes non-coding regions. Boxes with diagonal patterns indicate a homeobox domain (p.244–p.297). The missense variant, c.883A>C, is located in the homeobox domain.
|
Table 2
Pathogenic variants identified in patients with Currarino syndrome
All variants in the above eight patients are heterozygous.
*Only c.634C>T was previously reported [10], and the other variants were first reported in this study.
Abbreviations: ACMG, American College of Medical Genetics and Genomics; PVS, pathogenic very strong; PM, pathogenic moderate; PP, pathogenic supporting.
![]()
Pathogenic variants were more commonly identified in females (6/14) than in males (1/11). Of these, only one variant, c.634C>T (p.Gln212*), was previously reported in a Swedish patient [10]. The other variants were identified for the first time in this study.
Of the seven variants, p.Lys295Gln was the only missense variant. This variant was predicted to be “probably damaging” by Polyphen-2, “deleterious” by SIFT, and “disease causing” by Mutation Taster. In addition, this missense variant was located in a highly conserved homeobox domain between codons 244 and 297.
Almost all previously reported missense variants are located in the homeobox domain. Only a few of them (p.Met1Ile, p.Pro-27Leu, and p.Arg243Trp) have been reported outside of this region [182526]. Therefore, we classified p.Lys295Gln as a likely pathogenic variant. A synonymous variant, c.852G>A (p.Gln284=), was identified in patient 8. This variant was at the last nucleotide position of exon 2. The Human Splicing Finder predicted the wild type motif with a score of 87.86 and the mutant motif with a score of 77.28. The difference of the score between them (−12.04%) was under −10%, suggesting that the variant breaks the splice site. However, these findings were not sufficient to conclusively determine pathogenicity of the variant. Therefore, this synonymous variant was classified as a variant with uncertain significance.
We performed gene dosage analysis for 18 patients with negative results in the direct sequencing. MLPA analysis revealed a large deletion in patient 12 (Fig. 2). A reduced ratio was detected in all 11 probes in the 7q terminal region. This finding suggested that patient 12 had at least a 5.1-Mb 7q36 deletion. The exact breakpoints and range of the deletion could not be determined by MLPA results alone.
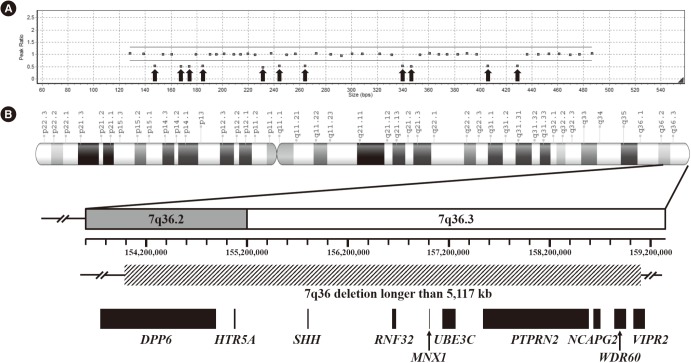 | Fig. 2Gene dosage analysis and mapping of the 7q36 deletion in patient 12. (A) Result of multiplex ligation-dependent probe amplification analysis. Arrows indicate the reduced ratio of all 11 probes in the 7q terminal region. These probes are located in MNX1, DPP6, RNF32, SHH, NCAPG2, WDR60, HTR5A, VIPR2, PTPRN2, UBE3C, and PTPRN2, in the order of increasing probe length. (B) Schematic representation of the 7q36 deletion. Genomic coordinates are based on GRCh38/hg38 assembly. Box with a diagonal pattern indicates a 7q36 deletion longer than 5.1 Mb. The exact breakpoints and range of the 7q36 deletion were not available. Black boxes indicate genes affected by 7q36 deletion.
|
3. Genotype-phenotype correlations
Seven of eight patients with pathogenic variants displayed the complete Currarino triad. Only three of 17 patients without pathogenic variants showed an incomplete Currarino triad without sacral anomaly. Patients with pathogenic variants presented predominantly with hemisacrum, anorectal stenosis, and teratoma for each Currarino triad. However, no significant phenotypic differences were observed between the patients with and without pathogenic variants. Patients in both groups exhibited variable expression of all the three major phenotypes and associated anomalies. The pathogenic variants were classified as six null variants, one missense variant, and one large deletion. The missense variant was associated with milder phenotypes (anorectal stenosis, cystic formation, and anterior angulation of coccyx) compared with phenotypes of the other null variants. The large deletion case presented with multiple associated malformations in addition to the classic CS triad.
A 3-year-old girl with suspected CS (Patient 1) was referred for genetic study. She did not excrete meconium within 24 hours of birth. Imaging studies suggested hemisacrum, high type imperforate anus, and teratoma. Pena operation and teratoma excision were performed at the age of five months. Direct sequencing revealed a frameshift variant, c.450_472del (p.Gly151Leufs*67), in the heterozygous state. She had no siblings. The patient's mother presented with fecal and urinary incontinence since birth. She underwent neurosurgical intervention for ligation of the anterior sacral meningocele and spinal cord untethering at the age of five years. After three months, Pena operation was performed to correct her low type imperforate anus. Imaging studies revealed sacral spina bifida and horseshoe kidney. There was no family history of CS in the patient's mother's parents or siblings. The same pathogenic variant as her daughter was identified by sequence analysis.
Go to :
DISCUSSION
The MNX1 sequence analysis identified six novel and one previously reported pathogenic variants. The overall detection rate of sequence analysis was 28% (7/25) in all patients and 25% (6/24) in sporadic patients. Haploinsufficiency of MNX1 causes CS; according to previous reports, pathogenic variants of MNX1 were detected in approximately 30% of CS sporadic patients [51718]. According to Cretolle et al [17], frameshift variants (45%) are the most frequent type of variants, followed by missense variants (25%) and nonsense variants (12%). Similarly, four frameshift variants, two nonsense variants, and one missense variant were identified in this study.
We identified a 7q36 deletion in a sporadic CS patient through gene dosage analysis. Large deletions or chromosome rearrangement involving 7q36 are related with holoprosencephaly (HPE) and CS due to haploinsufficiency of sonic hedgehog (SHH) and MNX1. Some previous studies reported patients with a deletion or chromosome rearrangement involving 7q36 [3414172728293031323334]. In these patients, microcephaly was the predominant craniofacial finding of HPE, and hypotelorism, ptosis, and midface hypoplasia were commonly associated. However, patients presenting with a complete CS triad were rare. Sacral anomaly was most frequently observed, followed by presacral mass and anorectal malformation. Pavone et al [3] described the first reported case with complete CS triad and microcephaly. A 10.3-Mb duplication of 7q34-q35 and an 8.8-Mb deletion on 7q36 were identified by comparative genomic hybridization (CGH) array in the patient. Another study reported a patient with a 2.7-Mb deletion 7q36.3 excluding SHH [28]. The patient presented with microcephaly and incomplete CS triad without anorectal malformation. In this study, we reported a patient with 7q36 deletion including loss of SHH and MNX1. This patient presented with complete CS triad and microcephaly, which was a rare phenotype spectrum upon 7q36 deletion.
CS shows characteristically diverse phenotypes with variable expressivity [51731], and there is no clear relationship between the genotype and phenotype. The 25 index patients revealed variable phenotypes in this study. Sacral anomalies varied from hemisacrum to partial sacral agenesis; likewise, presacral mass and anorectal malformation exhibited various pathological and clinical diagnoses. The clinical phenotypes were diverse, even in the familial case: patient 1 and her mother exhibited differences in all the three major phenotypes and their associated anomalies. Furthermore, phenotypic variability in familial cases has been previously reported [781011121831].
The fact that clinical variation was observed in individuals with the same variant suggests that factors other than MNX1 affect the CS phenotype. Other modifier genes might influence the MNX1 protein, partners, or transcriptional factors [5616]. Some candidates, such as proneuronal basic-helix-loop-helix proteins and LIM (named after Lin-11, Isl-2, and Mec-3 protein) domain proteins, have been found to regulate MNX1 expression in other vertebrates [353637]. A recent study using whole exome sequencing suggested three candidate genes, ETV3L, ARID3A, and NCAPD3, were involved in CS pathogenesis [38].
The most common malformations in patients with CS are urogenital anomalies and terminal spinal cord malformations. In this study, nine patients displayed these malformations. However, other associated malformations were also observed in some patients. Patient 12, in particular, with the complete Currarino triad, displayed severe associated malformations, including microcephaly and facial dysmorphism. Moreover, patient 16 also exhibited multiple associated anomalies including ptosis, bifid uvula, choanal atresia, laryngomalacia, Sprengel's deformity (congenital elevation of the scapula), cardiac anomalies, and umbilical hernia. According to the diagnostic algorithm proposed by Cuturilo et al [4], patients with CS exhibiting growth delay and/or developmental delay and/or facial dysmorphic features should be evaluated by a chromosomal microarray. Large deletions and complex rearrangements involving the 7q36 region have been identified in many previous reports [3414172728293031323334]. Almost all cases described in these studies exhibited various associated anomalies. In this study, we performed genetic analysis using direct sequencing and MLPA. Therefore, further studies using chromosomal microarray or whole exome sequencing will be needed to investigate patients with negative results, especially in cases with multiple associated malformations.
To our knowledge, only four studies have reported genetic analysis of a large series of patients with CS [5161718]. This study described the clinical and genetic aspects of 25 index patients with CS. We identified seven different pathogenic variants, six of which were novel, including a large deletion. The clinical variability of CS, including intrafamilial variability, was also confirmed. In conclusion, CS should be considered for early diagnosis even if the cases exhibit an incomplete CS phenotype. For adequate genetic counseling, genetic analysis should be considered for patients with suspected CS.
Go to :
 XML Download
XML Download