

 PDF
PDF ePub
ePub Citation
Citation Print
Print


Dear Editor,
Congenital analbuminemia (CAA; OMIM # 616000) is a rare autosomal recessive disorder characterized by the complete absence (or extremely low level) of serum albumin (ALB), and is usually diagnosed by serum protein electrophoresis [1]. Since different clinical conditions can result in hypoalbuminemia, mutation analysis of ALB gene is always necessary to confirm the diagnosis of CAA. Since the low ALB level is partially compensated by an increase of other serum proteins, CAA is considered a relatively benign condition in adulthood. Analbuminemic individuals do not usually show a severe phenotype and have few clinical symptoms [23]. The most common biochemical sign is hyperlipidemia, especially hypercholesterolemia and elevated LDL-cholesterol levels, reflecting the important role of ALB in lipid transport and metabolism [123]. However, CAA is a risk factor of high morbidity and/or mortality during pregnancy and childhood, suggesting that ALB plays a crucial role in the pre- and peri-natal period [45].
We investigated an unmarried 24-year-old woman and her father from Tissemsilt (Northern Algeria), and two young sisters and their parents from Antalya (Southern Anatolia, Turkey). This study was exempted from ethical approval. After obtaining informed consent, whole blood samples were collected from the subjects for biochemical and genetic studies. Diagnosis and biochemical assessments for the Algerian family members were performed at the Hemobiology Center- Blood Transfusion Center and the Laboratory of Medical Virology, Mustapha Bacha University Hospital Center, Algiers, between October 2014 to January 2015.
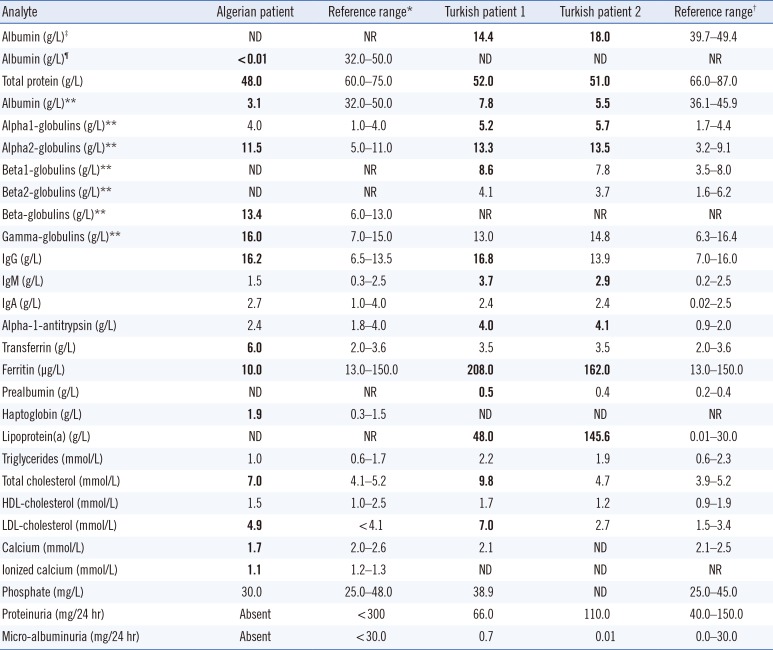
At diagnosis, the proband showed edema in the lower limbs lasting for six months, without any benefit from specific treatment. Her parents are first-degree cousins, and her sister had died in her first year of life. Conventional serum protein electrophoresis showed hypoalbuminemia, and an immunonephelometric assay revealed the nearly total absence of ALB; her lipid profile demonstrated increased total and LDL-cholesterol levels (Table 1).
Diagnosis and biochemical analysis for the members of the Turkish family were carried out at the Department of Pediatric Gastroenterology, Akdeniz University, Antalya, Turkey in 2014. In this case, the parents are first-degree cousins. One of the sisters (patient 1) was 14-year old at the time of diagnosis. She had no complaints apart from obesity and some enlarged varicose veins affecting her legs. A modified bromocresol green-binding assay revealed marked hypoalbuminemia, which was confirmed by capillary electrophoresis. She had gross hypercholesterolemia with a significant increase of the LDL fraction (Table 1). The main clinical symptoms of her 11-year-old sister (patient 2) were light edematous face and slight pre-tibial edema. She also showed marked hypoalbuminemia. Her lipid profile was essentially normal, except for a significant increase of the Lp(a) level (Table 1), suggesting that the biochemical signs may be ambiguous among individuals affected by CAA.
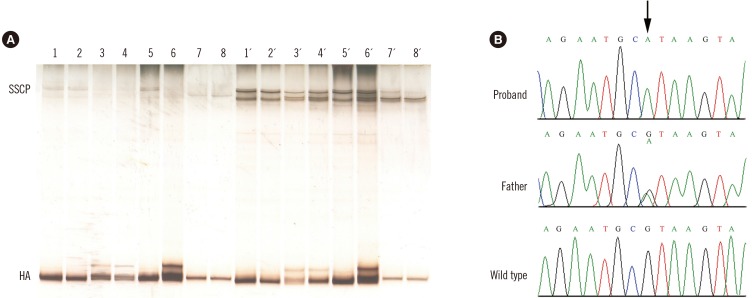
The wide range of ALB levels for the three probands may be explained by the different quantification methods employed. The dye-binding technique used for the two Turkish sisters suffers from poor accuracy in cases of low ALB levels, as this method largely overestimates the true amount in these conditions. In contrast, ALB immunoassay used for the Algerian patient is the most accurate technique available [26]. To analyze the causative molecular defects in these cases, we used our previously reported strategy for rapid identification of mutations in ALB (GenBank mRNA refseq: NM_000477.6) [7]; it is based on PCR amplifications described by Watkins et al [4], followed by heteroduplex analysis (HA) and single-strand conformational polymorphism (SSCP) analysis. Both HA and SSCP analysis revealed that all three probands were homozygous and all the examined parents were heterozygous for the same mutation in the PCR fragment encompassing exon 10 and flanking intronic junctions [7] (Fig. 1A). The electrophoretic behavior of the abnormal PCR fragments is essentially identical to that previously observed in a Portuguese boy affected by CAA and named analbuminemia Guimarães [8]. DNA sequence analysis of this region confirmed that all three probands were homozygous for a G>A transition at nucleotide c.1289+1, the first base of intron 10 (Fig. 1B), and all the examined parents were heterozygous for the same mutation.
After our study of these three new cases, the splicing defect of analbuminemia GuimarÃes now represents the second most common cause of CAA, accounting for four among the 43 cases molecularly characterized to date [3]. The most common molecular defect is the deletion of two bases c.228_229delAT (analbuminemia Kayseri), which has been the cause of 13 CAA cases [3]. Our report of the Guimarães transition in apparently unrelated, and geographically distant, families suggests that the sequence CpG at the 5´ end of intron 10 may represent a mutation hot-spot in ALB. Close follow-up of subjects with analbuminemia, together with identification of molecular defects causing new possible CAA cases, will be necessary to better elucidate the phenotype and molecular genetics underlying this condition.
 XML Download
XML Download