

 PDF
PDF ePub
ePub Citation
Citation Print
Print


INTRODUCTION
Rhabdomyosarcoma (RMS) is the most common soft tissue sarcoma in children,accounting for 6–8% of all childhood malignancies. RMS is classified into two major histopathological subtypes, alveolar RMS (ARMS) and embryonal RMS (ERMS). ERMS accounts for 70% of RMS cases and is associated with a relatively favorable prognosis. ARMS comprises 20% of RMS cases and has a more aggressive behavior, with early dissemination, poor response to treatment, and frequent relapses following treatment [1234]. Clinical symptoms of RMS are associated with the primarily affected organs. For example, frequent urination can be an initial presentation of RMS that arises within the urinary bladder. Obstructive jaundice is one manifestation of bile duct RMS [4].
ERMS usually occurs in children younger than 15 years. It is characterized by complex genetic changes involving various chromosomal gains and losses. However, these abnormalities are not specific to the disease [56]. ARMS exhibits typical cytogenetic characteristics, including t(2;13)(q35;q14) or t(1;13)(p36;q14), which result in PAX3-FOXO1 or PAX7-FOXO1 fusion genes. The t(2;13)(q35;q14) and t(1;13)(p36;q14) have been detected in 55% and 22% of ARMS cases, respectively [7]. Detection of the fusion gene by RT-PCR or of the FOXO1 translocation by FISH is commonly used for molecular diagnosis [8]. Approximately 20% of RMS patients present with metastases at the time of diagnosis; lung, bone, and bone marrow are frequently involved. A previous study suggested that patients with fusion transcripts or myoD1 transcripts in the bone marrow or peripheral blood are at high risk of tumor progression [9]. However, the clinical impact of chromosomal abnormalities in the bone marrow has yet to be comprehensively examined.
In the present study, we analyzed the clinical and laboratory data of childhood RMS patients at a single institution and determined the cytogenetic profiles and clinical outcomes according to bone marrow involvement and chromosomal abnormalities.
MATERIALS AND METHODS
1. Patients
A total of 51 Korean patients with childhood RMS were enrolled in this retrospective study. All patients were younger than 18 years at diagnosis and were treated at Asan Medical Center, Seoul, Korea, between 2001 and 2015. The median age of all patients was six years (range, 0–18 years), and the number of males and females were 32 and 17, respectively. The medical records were reviewed to identify clinical features, laboratory data, staging, treatment, and outcome. Staging was determined using the Intergroup Rhabdomyosarcoma Study (IRS) TNM classification for pretreatment clinical assessment of disease [4]. RMS bone marrow involvement was defined primarily based on the morphological characteristics of neoplastic cells. Neoplastic cells showed a medium to large size, oval nucleus, and a limited or moderate amount of blue cytoplasm. In cases with morphological ambiguity, additional immunohistochemistry stains, involving the desmin, myogenin, CD56, and CD99 antibodies, were performed. The study was approved by the Institutional Review Board of Asan Medical Center (2017-1230).
2. Chromosomal analysis and FISH
Chromosomal analysis was performed on bone marrow specimens, which were collected at diagnosis from 40 patients using short-term cultures and the Geimsa-Trypsin-Leishman (GTL) banding technique. The karyotype was described according to the International System for Human Cytogenetic Nomenclature (ISCN 2013). FISH was conducted with the bone marrow specimens using a FOXO1 break-apart probe (Abbott Laboratories, Abbott Park, IL, USA) in three patients and a MYCN/CEP2 probe (Abbott Laboratories) in one patient with double minute chromosomes. FISH procedures were performed according to the manufacturer's instructions and only for four patients, because we could not obtain fresh specimens from patients who were diagnosed as having RMS before 2014.
3. Statistical analysis
Categorical variables were compared using the Fisher's exact test, and continuous variables were compared using the Mann-Whitney U-test. Overall survival (OS) was calculated from the time of diagnosis to death due to the disease or the last follow-up. The Kaplan-Meier method was applied to generate survival curves for OS, and the log-rank test was used to compare survival across groups. Results of multivariate analyses of the different prognostic factors (e.g. disease type, stage, bone marrow involvement, or chromosomal abnormality) were evaluated using the Cox proportional hazards regression model. Hazard ratios (HRs) and their 95% confidence intervals (CIs) were obtained. Statistical significance was defined as P<0.05. Statistical analyses were performed with SPSS software version 18.0.0 (SPSS, Chicago, IL, USA).
RESULTS
1. Patient characteristics
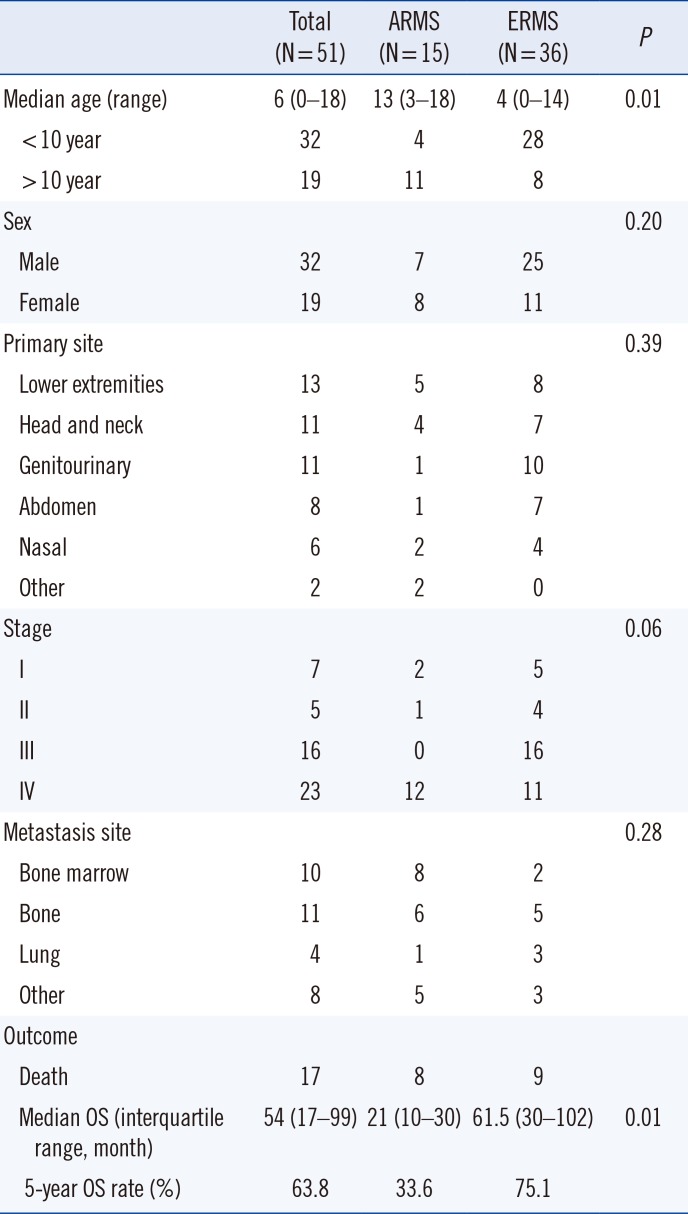
Of the 51 study patients, 36 (70.6%) had ERMS and 15 (29.4%) had ARMS. The median age of ARMS was 13 years (range, 3–18 years) and that of ERMS was four years (range, 0–14 years) (P=0.01). The sex ratio was 1.7:1 for all patients (male:female), 0.9:1 for the ARMS patients, and 2.3:1 for the ERMS patients (P=0.2). The primary sites involved were diverse, and the lower extremities, including the thigh and buttock, were the sites most commonly involved. The head, neck, and genitourinary sites were the second most commonly involved sites. Genitourinary and abdominal sites were more frequently involved in ERMS. The proportion of patients with stage IV disease was significantly different between ARMS and ERMS (80% in ARMS vs 31% in ERMS, P=0.02). Eleven patients (21.6%) had bone metastases, 10 (19.6%) had bone marrow metastases, and four (7.8%) had lung metastases. The clinical findings of the ARMS and ERMS patients are summarized in Table 1.
2. Bone marrow involvement and cytogenetic findings
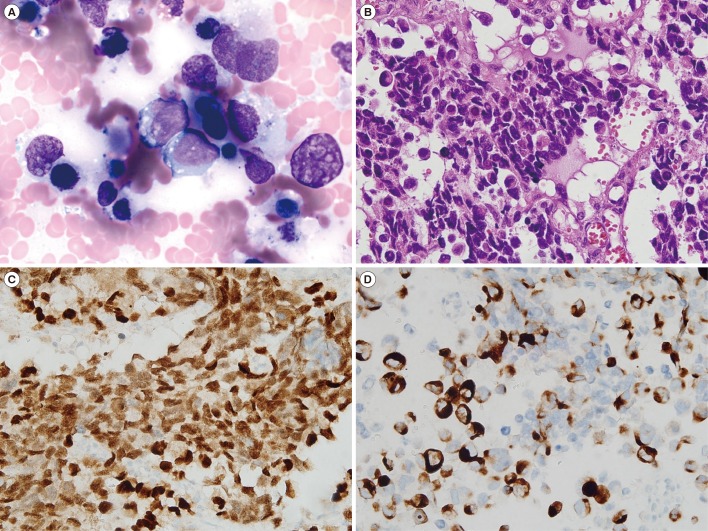
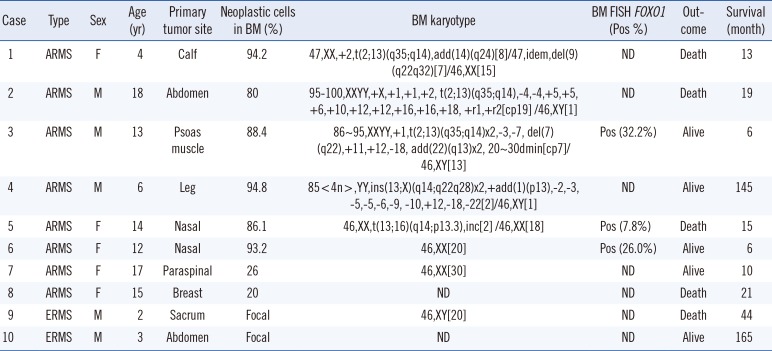
The characteristics of the 10 patients with bone marrow involvement are outlined in Table 2. Metastasis to the bone marrow was more frequent in ARMS (53.3%, 8/15) than in ERMS (5.6%, 2/36; P<0.01). Six of the eight ARMS patients with bone marrow involvement showed massive infiltration of >80% neoplastic cells (Fig. 1A and 1B), whereas the two ERMS patients with bone marrow involvement showed a focal involvement pattern. Immunohistochemical staining was performed with the desmin, myogenin, CD56, and CD99 antibodies. Most cases were positive for myogenin and desmin (Fig. 1C and 1D).
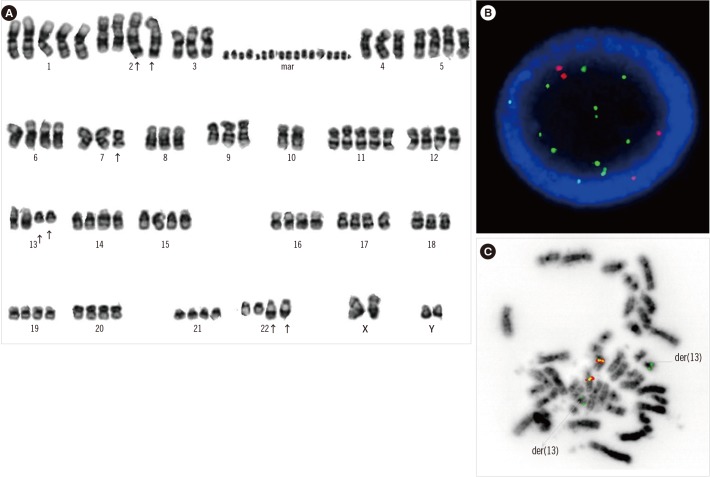
Of the 51 patients, we could analyze cytogenetic profiles for 40 patients; among them, 32 patients showed no evidence of bone marrow involvement, and eight patients showed bone marrow involvement of RMS. The patients without bone marrow involvement all showed a normal karyotype. Five patients with bone marrow involvement exhibited chromosomal abnormalities. All five patients had ARMS and showed massive infiltration of the bone marrow. Three of these cases were associated with t(2;13)(q35;q14), whereas the other two had ins(13;X)(q14;q22q28) and t(13;16)(q14;p13.3), respectively. Three patients had a near-tetraploid modal number with complex chromosomal abnormalities. FOXO1 FISH was performed in three ARMS patients (patients 3, 5, and 6), and all patients were positive for FOXO1 rearrangement. One patient (patient 3) with a t(2;13) and numerous double minute chromosomes had a FOXO1 rearrangement and MYCN amplification (Fig. 2). Unfortunately, FOXO1 FISH was performed for only three cases with bone marrow involvement, which constitutes a limitation of this study.
3. Clinical outcomes
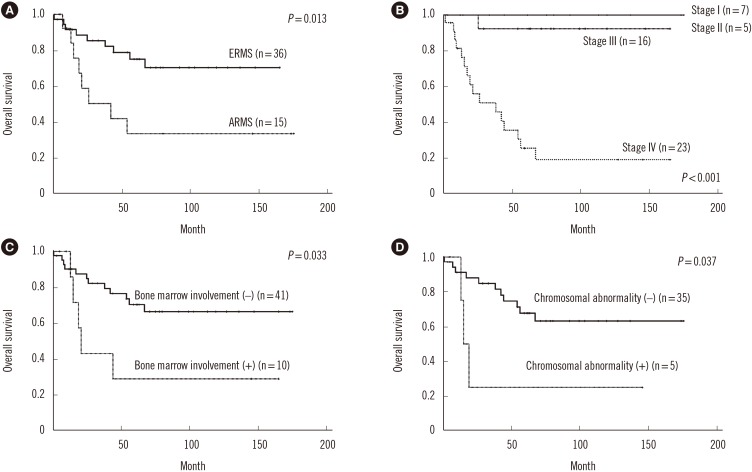
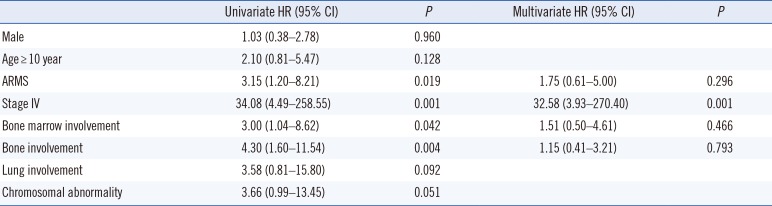
The 5-year OS rate was 63.8% in all RMS patients, 33.6% in ARMS patients, and 75.1% in ERMS patients. The median OS was 54 months (range, 1–175 months) and 17 patients died. ARMS patients had a markedly shorter median OS than ERMS patients (21 vs 62 months, P=0.013; Fig. 3A). Patients with stage IV disease had a significantly poorer OS than those with other stages (Fig. 3B). A significantly shorter OS was also observed in patients with bone marrow involvement (17 vs 61 months, P=0.033) and with a chromosomal abnormality (15 vs 61 months, P=0.037; Fig. 3C and 3D). A non-significant, shorter OS was observed among ARMS patients with bone marrow involvement (14 vs 54 months; P=0.20) and those with chromosomal abnormalities (15 vs 42 months, P=0.30). Multivariate analysis indicated that stage IV disease was significantly associated with a poorer OS (HR, 34.21; 95% CI, 4.36–268.58; P=0.001) (Table 3).
DISCUSSION
RMS is a relatively rare cancer; however, it is the most common soft tissue sarcoma in children and young adults, accounting for over 50% of soft tissue sarcomas in these age groups [1011]. The proportions of ARMS and ERMS in this study were 29.4% and 70.6%, respectively, which are similar to the results from the Children's Oncology Group and a large Japanese cohort [1213]. However, the stage distribution in our study was different from that of previous studies, with a higher percentage of stage IV patients (45.1%) than the Children's Oncology Group (16%) and the Japanese group (23.2%). Stage IV disease was observed in 80% of ARMS patients and 31% of ERMS patients.
The frequency of metastatic involvement of the lung, bone, and bone marrow in RMS patients was 6%, 5%, and 6%, respectively, according to data from the Children's Oncology Group. The frequencies of bone, bone marrow, and lung metastases in the current study were similar to those observed in other studies. In particular, 53% and 40% of the ARMS patients in the current study had bone marrow and bone metastases, respectively.
In a previous report, the estimated 5-year OS rate was 47.8% for patients with ARMS and 73.4% for patients with ERMS from 1996 to 2000 [14]. Our data showed similar results for ERMS, but a lower 5-year OS rate for ARMS. ARMS patients with bone marrow involvement and chromosomal abnormalities appeared to show a trend towards poorer clinical outcomes than patients without these features.
The molecular characteristics of ARMS are thought to be mediated by PAX3-FOXO1 or PAX7-FOXO1 fusion genes. The tumorigenic mechanism of these fusions is not well understood; however, a recent study showed that PAX3-FOXO1 may contribute to tumor formation by inhibiting the tumor-suppressor activities characteristic of both FOXO family members and transforming growth factor beta (TGF-β) pathways [15]. Other gene rearrangements have also been described in ARMS patients. These include t(2;X)(q35;q13), which results in a gene fusion of PAX3 with AFX [16], and t(2;2)(q35;p23) and t(2;8)(q35;q13), which generate the PAX3-NCOA1 and PAX3-NCOA2 fusion proteins, respectively [17]. In our study, most of the ARMS patients with bone marrow involvement had FOXO1-related chromosomal abnormalities. Importantly, we identified two novel rearrangements associated with the 13q14 locus, ins(13;X)(q14;q22q28) and t(13;16)(q14;p13.3). To the best of our knowledge, these rearrangements have not previously been described. The t(13;16) anomaly was revealed to result in FOXO1 rearrangement by FOXO1 FISH analysis. Unfortunately, we were unable to identify the partner genes of FOXO1.
Previous reports using spectral karyotype analysis identified chromosomes 1, 3, and 13 as the chromosomes of origin of the double minute chromosomes [6]. Here, we found an MYCN amplification in one patient with numerous double minute chromosomes. ARMS with MYCN amplification was associated with higher stage disease, larger tumor, metastatic disease, and inferior survival [2]. Our results are consistent with these findings, because our patient was classified with stage IV disease, and the presence of multiple metastases was determined by radiology.
This study had some limitations. First, our study had a smaller sample size than other studies. Therefore, additional large-scale domestic research is needed in the future. Second, we did not perform FOXO1 FISH on all specimens from patients with bone marrow involvement. In summary, we describe the clinical features, cytogenetic characteristics, and novel chromosomal rearrangements of Korean RMS patients. Our results indicate that chromosomal investigation of the bone marrow could have prognostic significance and provide a better understanding of the cytogenetics of RMS, such as novel fusion genes and MYCN amplification. In addition, even with a limited number of patients, bone marrow involvement and chromosomal abnormality were shown to be related to shorter OS. Therefore, we suggest a comprehensive approach involving conventional cytogenetics, FISH, histopathology, and immunohistochemistry for assessing high-risk ARMS patients, and a multi-center study will be needed to clarify the clinical and cytogenetic profiles more accurately.
 XML Download
XML Download