

 PDF
PDF ePub
ePub Citation
Citation Print
Print


INTRODUCTION
Increasing data have demonstrated that colonizing microorganisms might dictate human health and disease [1]. Although the 2012 Human Microbiome Project did not include the lower respiratory tract in its surveys, advances in sequencing technology have revealed a diverse population of non-routinely cultured bacteria in the lung, which has traditionally been thought to be sterile [2345]. Most of these studies are based on 16S rRNA next generation sequencing (NGS), a highly sensitive method for analyzing the microbiome [6]. Because the human microbiota consists of many ‘unculturable’ bacteria, culture-independent metagenomic approaches have driven significant advances in microbiome research. However, the bacterial species information obtainable from these metagenomic approaches might be limited, and there are several steps, including nucleic acid extraction, PCR amplification, sequencing (method and depth), and bioinformatic analysis, that may produce variations in the results. Read length is also one of the most important factors when considering clinical application. Hence, the cultivation of microbes provides more reliable information regarding the phenotype and ecological role, allowing a greater understanding of microbe-host interactions [7].
Recently, a number of new approaches for gut microbiota cultivation have emerged with potential for use in microbiome research [8]; these have mainly been applied to investigate the gut microbiome [91011]. Although these novel approaches have shown that culture can capture a significant subset of the species identified using culture-independent methods [8], it is not easy to apply these techniques in clinical laboratories. In addition, limited information is available regarding culture-based microbiome analysis of the respiratory tract compared with that of the gut. We used culture conditions commonly adopted in conventional clinical microbiology laboratories to explore the micro-biota in bronchoalveolar lavage (BAL) fluid compared with the microbiome results obtained via 16S rRNA NGS.
METHODS
1. Sample collection
BAL fluid samples were collected from patients undergoing bronchoscopy procedures for lung mass evaluation at Severance Hospital, Seoul, Korea from May to November 2015. The bronchoscope was inserted through the mouth, and BAL was collected according to a standardized protocol at the opposite side of the lung mass; 10 mL BAL fluid was acquired from the patients with approximately 30 mL sterile 0.9% saline. Sample was collected in accordance with the Institutional Review Board of Severance Hospital (IRB No. 4-2014-1014); informed consent was obtained from the patients.
2. Culture conditions
BAL fluid samples were cultured on the day within 6 hours of collection. Undiluted BAL fluid (100 µL) was inoculated onto an agar plate using a spreader. Different agar plates with aerobic and anaerobic conditions were used: blood agar (aerobic, 35℃, 5% CO2), chocolate agar (aerobic, 35℃, 5% CO2), MacConkey agar (aerobic, 35℃, 5% CO2), Brucella agar (anaerobic, 35℃), phenylethyl alcohol agar (anaerobic, 35℃), and Sabouraud dextrose agar (aerobic, 30℃, ambient air). Bacterial colonies were isolated after 48 hours of culture. Sabouraud dextrose agar (SDA) plates used for fungal culture were observed for up to four weeks of incubation.
3. Bacterial identification
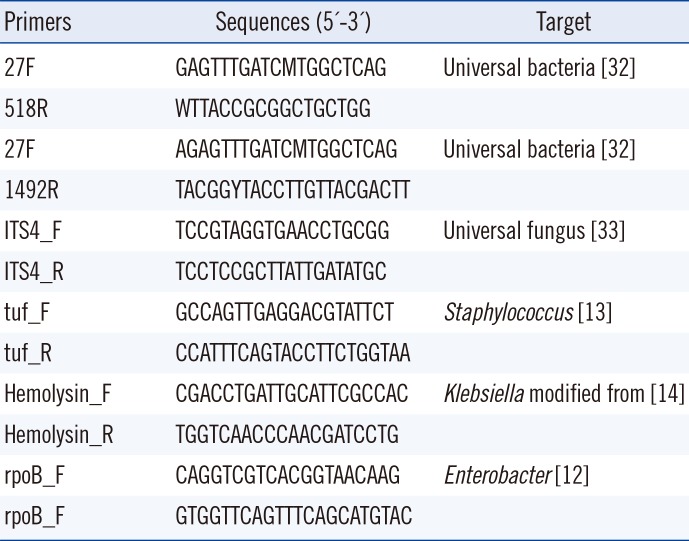
An average of 20 colonies with different morphologies and sizes were selected from each agar plate and applied to a Microflex LT matrix-assisted laser desorption/ionization time-of-flight mass spectrometry (MALDI-TOF MS) system with Biotyper software 3.1 (Bruker Daltonics, Bremen, Germany) for simultaneous identification and subculture. When an accurate identification was not available (score <1.7) using only α-cyano-4-hydroxycinnamic acid, formic acid was added. Strains that could not be reliably identified by MALDI-TOF MS were subjected to further analysis; the subcultured colonies were analyzed using 16S rRNA sequencing for bacteria and ITS sequencing for fungi performed with primers 27F-1492R and ITS4, respectively (Table 1). To detect Staphylococcus spp., Klebsiella spp., and Enterobacter spp. in samples from which bacterial species were isolated solely on culture plates, we performed PCR amplification using organism-specific primers as previously described [121314] (Table 1).
4. High-throughput sequencing
DNA was extracted from BAL fluid within 6 hours of sample collection by using the DNeasy Blood & Tissue kit (Qiagen, Venlo, Limburg, Netherlands), according to the manufacturer's protocol. DNA extraction was performed simultaneously with bacterial culture. The DNA samples were stored at –70℃ until further analysis. The 16S rRNA gene from each DNA sample was amplified using barcoded fusion primers targeting the V1 to V3 regions (27F-518R), because this region has been reported to be reliable for accurate taxonomic classification of bacterial sequences [15] (Table 1). Amplification was carried out under the following conditions: initial denaturation at 95℃ for 5 minutes; followed by 30 cycles of denaturation at 95℃ for 30 seconds, primer annealing at 55℃ for 30 seconds, and extension at 72℃ for 30 seconds; and a final elongation step at 72℃ for 7 minutes. PCR products were confirmed using 2% agarose gel electrophoresis and visualized with a Gel Doc system (BioRad, Hercules, CA, USA). The amplified products were purified using the QIA-quick PCR purification kit (Qiagen). Equal concentrations of purified products were pooled together, and short fragments (non-target products) were removed using the AMPure beads kit (Agencourt Bioscience, Beverly, MA, USA). Quality and product size were assessed on a Bioanalyzer 2100 (Agilent, Palo Alto, CA, USA) using a DNA 7500 chip. Mixed amplicons were obtained by emulsion PCR and then deposited on picotiter plates. Sequencing was carried out at Chunlab, Inc. (Seoul, Korea) using the GS Junior Sequencing system (Roche, Branford, CT, USA) according to the manufacturer's instructions. Reads obtained from different samples were sorted using the unique barcodes on each PCR product. The barcode, linker, and primer sequences were removed from the original sequencing reads. Any reads containing two or more ambiguous nucleotides, low-quality score (average score <25), or reads shorter than 300 bp were discarded. Potential chimera sequences were detected using the UCHIME algorithm [16]. Following removal of the chimera sequences, the taxonomic classification of each read was assigned against the EzTaxon-e database (http://eztaxon-e.ezbiocloud.net) [17].
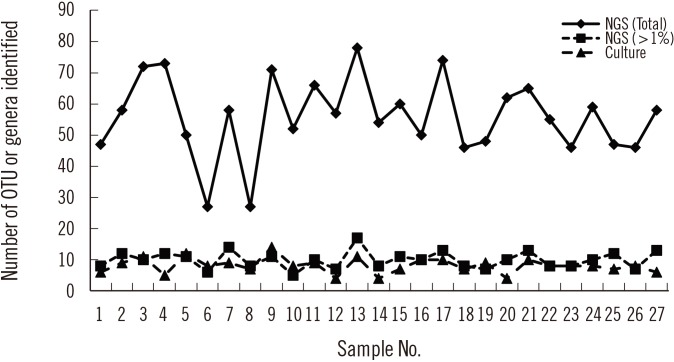
The operational taxonomic units (OTUs) identified at the genus level were listed with the number of reads, reflecting the relative frequency of detection. Next, the number of cultured colonies was compared with the number of OTUs determined by NGS in the same sample to analyze the difference in identification by NGS and culture.
RESULTS
1. Culture and MALDI-TOF MS identification
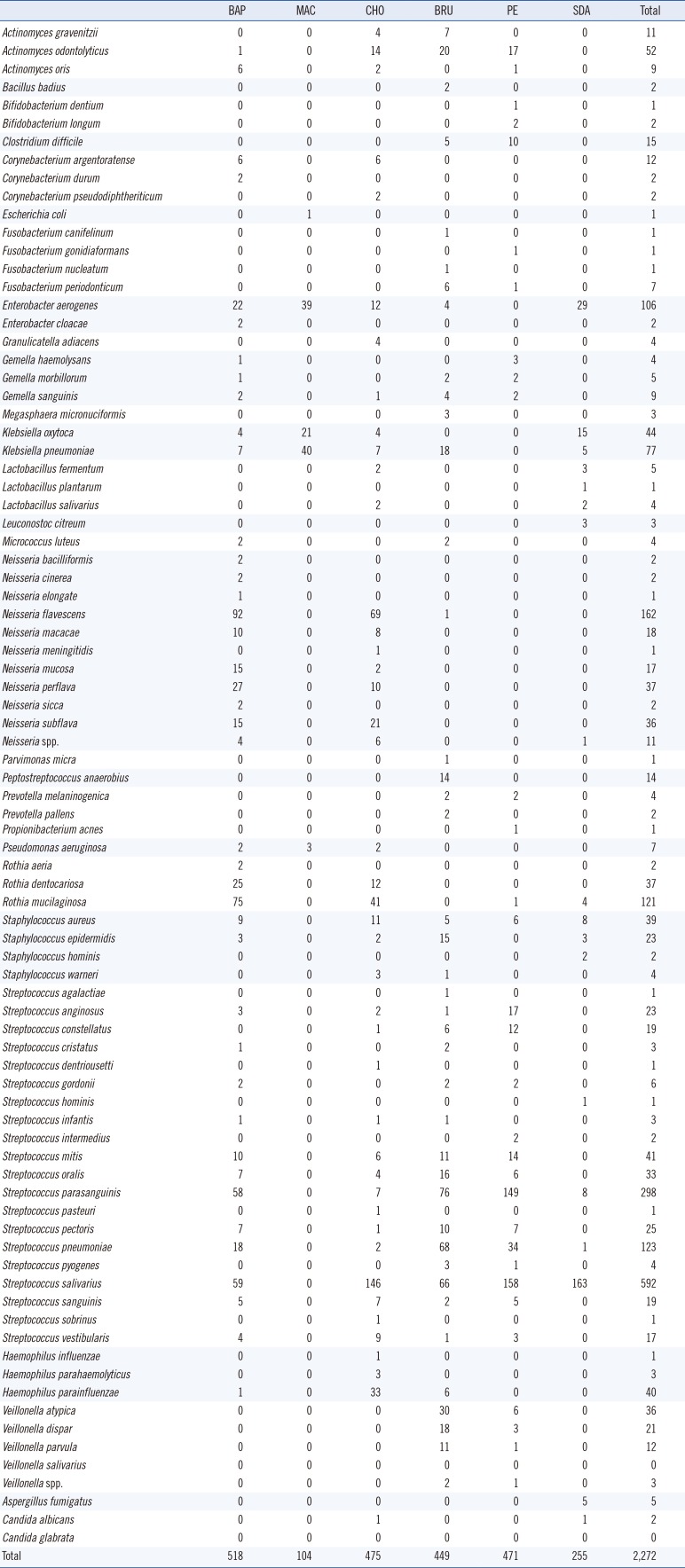
A total of 2,272 microbial colonies were isolated from 27 BAL fluid samples and identified using culture and MALDI-TOF MS. The number of genera identified per sample ranged from 4 to 14, with an average of 8. The total number of genera identified was 29; these included 85 species. Streptococcus spp. were the predominant isolates with a total number of 1,213 colonies (Table 2). Culture on blood and MacConkey agar resulted in the highest and the lowest number of microorganisms, respectively. Forty (1.76%, 40/2,272) colonies were not identified by MALDI-TOF MS. These colonies were subcultured and re-identified using MALDI-TOF MS with the aid of formic acid, yielding mainly Streptococcus spp. (47.5%, 19/40). Four colonies (0.18%, 4/2,272) could not be identified with repeated MALDI-TOF MS; these were identified as Actinomyces odontolyticus, Actinomyces gravenitzii, Fusobacterium periodonticum, and Aspergillus fumigatus by sequencing using 16S rRNA and ITS gene amplification.
2. 16S rRNA NGS identification
Sequencing using the GS Junior Sequencing system generated 10,332 valid reads per sample on average (range 6,761–19,764). At the genus level, a total of 288 OTUs were identified with a mean of 55.8 (range 27–78) per sample. Considering those with a relative abundance greater than 1%, the mean number of OTUs identified at the genus level was 10.0 (range 6–17) per sample (Fig. 1), which is comparable to the number of bacterial genera identified using conventional culture and MALDI-TOF MS. Prevotella spp. was the most frequent and abundant OTU identified at the genus level; it was identified in all 27 samples (relative abundance 6.5–54.5% with a mean of 29.2%). The next most prevalent OTUs (mean relative abundance) were Streptococcus (11.5%), Neisseria (10.5%), Haemophilus (10.1%), and Veillonella (9.1%).
3. Comparison of microbial identification using culture and MALDI-TOF MS vs NGS
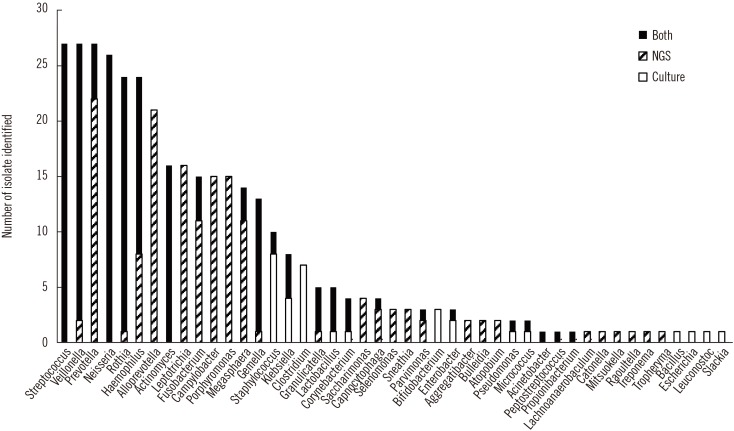
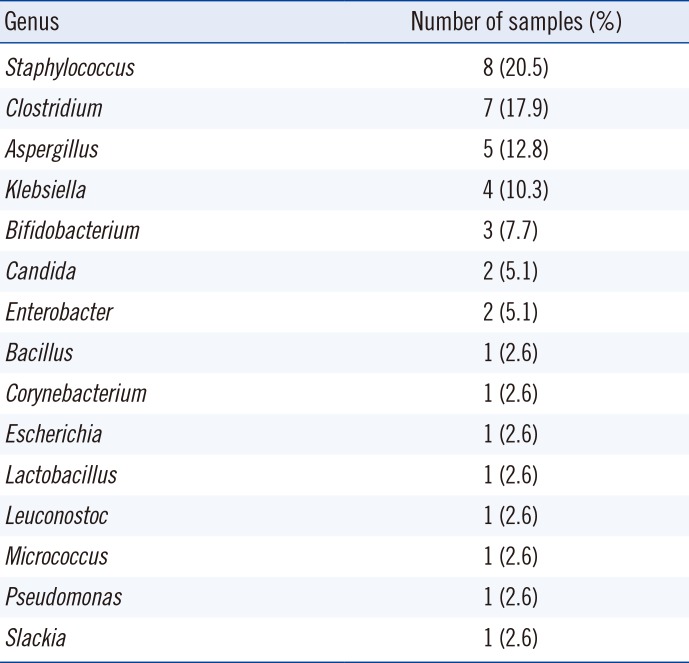
NGS revealed more OTUs compared with the number of genera identified by culture (Fig. 1). The relative abundance of bacterial reads identified simultaneously by culture was 47.1% on average, ranging from 3.2% to 88.0% in the 27 samples. The bacteria present in BAL fluids according to the identification method (NGS, culture, or both) are shown in Fig. 2. Of the 156 bacterial genera identified solely by NGS with >1% relative abundance, Prevotella spp. was the most common (14.1%) and frequent (22 of 27 samples, 81.5%) genus. When considered as relative abundance, the genus Prevotella spp. comprised over 50% of the bacteria that were sequenced but not cultured. Interestingly, some bacteria were identified solely by culture; of these, Staphylococcus spp., Clostridium spp., and Enterobacteriaceae including Klebsiella spp. were common, comprising 66.7% (18/27) of the 27 samples and 56.4% (22/39) of the total number of colonies that were cultured but not sequenced (Table 3). We per-formed organism-specific PCR using the primers detailed in Table 1 to detect Staphylococcus spp., Klebsiella spp., and Enterobacter spp. All PCRs failed to amplify the target organisms in the samples, suggesting DNA instability in clinical samples. Candida and Aspergillus spp. were only cultured because they cannot be identified by 16S rRNA PCR.
DISCUSSION
The purpose of this study was to evaluate the ability of the standard culture identification method used in clinical microbiology laboratories to elucidate microbial community profiles in BAL fluids. We compared the BAL fluid microbiota profiles identified by two methods: culture with MALDI-TOF MS and NGS. As expected, we failed to detect many strict anaerobic bacteria with the culture method. The most common bacteria identified solely by 16S rRNA NGS were Prevotella spp., which are abundant in respiratory specimens [18]. In a previous study, Prevotella spp. were the most abundant OTUs in BAL fluid followed by Streptococcus spp. and Neisseria spp. [19]. Prevotella spp. were isolated on Brucella or PE agar in only five samples, although sequencing identified this genus in all 27 samples. Interestingly, positive correlations between bacterial spp. and culture media were noted. For example, Streptococcus spp. grew prominently in chocolate agar under 5% CO2, and Brucella agar under anaerobic conditions. In addition, it is difficult to determine whether some of the OTUs detected solely by NGS might be contaminants [20].
Other bacteria that could not be cultured included many anaerobes such as Porphyromonas spp. and Leptotrichia spp. Failure of anaerobic culture is the most likely reason, although the viability of the sequenced bacteria without growth in culture media must be considered. It is interesting that despite the limitations of the culture conditions adopted in this study, some bacterial species, including Staphylococcus spp., Clostridium spp., and Bifidobacterium spp. were cultured, but not sequenced. Selective media (MacConkey agar and PE agar) and enriched media (chocolate and Brucella agar) were successful in identifying bacteria that were not detected using NGS. We hypothesize that culture can be more sensitive than sequencing in terms of detection under certain conditions and for some bacterial species, as shown in previous studies for Staphylococcus aureus [10] and Bifidobacterium spp. [21]. We assume that the bacteria identified solely by culture existed in very low quantities and thus failed to be sequenced. Another possible explanation is that the growth of some bacteria may be affected by culture media in association with other co-existing bacteria. The use of mixed culture via co-cultivation with helper strains could be used to facilitate growth [22 23]. As previously reported, the information provided by metagenomic approaches can be limited [24]. Many pre-analytical, analytical, and post-analytical factors can affect the outcome. DNA extraction is another step that can influence sequencing results [252627]. DNA isolation methods, the 16S rRNA target region used for PCR, and sequencing vary widely, and differences in techniques and manufacturers' kits may produce disparate data [28]. In this study, the V1-V3 region (27F-518R) of the 16S rRNA gene was amplified, because it is one of the most reliable targets for taxonomy. However, a sequencing depth of 10,332 valid reads per sample on average could affect the identification results. In addition, the culture method enabled the identification of fungi, such as Candida and Aspergillus, which cannot be detected by 16S rRNA sequencing.
It is known that a large proportion of the microbes found in the human gastrointestinal tract are unculturable [29 30], although this has been challenged by recent studies revealing many rare microorganisms. Metagenomics by deep sequencing is undoubtedly a powerful tool in microbial community analysis; however, it possesses some limitations. Sequenced DNA is not evidence of viable bacteria, and detection sensitivity is dependent on sequencing depth, which may result in a failure to identify low-abundance bacterial species. NGS approaches, especially those based on amplification of the highly conserved 16S rRNA gene, are inherently incapable of detecting intra-species variations [24]. Cultivation of microbes provides a greater understanding of bacterial interactions and traits and can be used to obtain completely sequenced genomes from these species. In addition, currently, the culture method with MALDI-TOF MS has advantages over NGS, in terms of cost and practical application. Although NGS has reduced the cost of sequencing greatly, it is still a fairly expensive technology and requires bioinformatic tools for analysis.
Recent efforts in developing culture methods have enabled the culture of previously ‘as-yet uncultured’ bacteria, known as culturomics [10, 3132]. The use of several different culture conditions has enabled the isolation of new bacterial species, the largest human virus, the largest bacterium, and the largest archaeon from humans [33]. One study demonstrated that analyses of the same samples produced only partially overlapping results; 51 of the 340 species and 698 phylotypes were identified by culturomics and metagenomic analysis, respectively [10]. However, various culture conditions, including many nonselective and selective culture media used in culturomics, are not practical for application in conventional clinical microbiology laboratories. MALDI-TOF MS is another powerful tool used in microbial identification. MALDI-TOF MS has allowed for the accurate identification of a large panel of anaerobes that are poorly identified because of ambiguous or erroneous results using conventional methods. Coupling MALDI-TOF MS with culturomics has enabled the identification of a large collection of bacterial species, including anaerobes that are usually poorly identified with current phenotypic methods, from stool specimens [34]. When a limited number of culture conditions commonly used in clinical microbiology laboratories were applied, all the isolated bacteria were present in the MALDI-TOF MS database, although some bacteria were poorly identified and needed further 16S rRNA or ITS sequencing.
Our study has some limitations. We used limited types of culture media commonly used in clinical microbiology laboratories. While picking the colonies from each agar plate, some bacteria may have been missed because of selection bias. The agar plates were incubated for only 48 hours; this duration may not have been adequate for slow-growing microorganisms. Although we tried to inoculate the samples as soon as possible and within 6 hours post collection, some anaerobic bacteria may have been lost during the prolonged oxygen exposure. The number of reads, ~10,000 per sample on an average in this study, may also affect the bacterial species detected in each sample. Most of the bacterial species were successfully identified using MALDI-TOF MS. However, the reliability of streptococci identification might not be high because of the inherent limitations of MALDI-TOF MS. Lastly, we did not quantify the microorganisms in in culture method, which is a limitation in exploring microbial diversity.
To the best of our knowledge, this is the first study to evaluate the use of conventional culture methods combined with MALDI-TOF MS in microbiome analysis of BAL fluid samples compared with 16S rRNA NGS. Some bacteria, mainly anaerobes, failed to grow on conventional culture media, and less diverse microbial communities were identified with the culture method. However, interestingly, some common bacteria were not identified by sequencing, but were cultured, indicating the significance of culture in microbiome analysis. Despite a number of limitations, culture methods with MALDI-TOF MS may play a significant role in microbiome analysis that is complementary to 16S rRNA NGS.
 XML Download
XML Download