

 PDF
PDF ePub
ePub Citation
Citation Print
Print


Dear Editor,
Acromicric dysplasia (AD, MIM 102370) is a rare skeletal dysplasia characterized by severe short stature, short hands and feet, normal intelligence, and mild facial dysmorphism [1]. The radiological manifestations of AD include delayed bone age, cone-shaped epiphyses, internal notch of the femoral head, short metacarpals, and short phalanges [2]. Recently, missense mutations clustered in exons 41 and 42 in the transforming growth factor-β binding-protein-like domain 5 (TB5) of FBN1 were identified as causative mutations for AD [23]. Here, we present the clinical and radiographic findings and effects of recombinant human growth hormone (rhGH) treatment for 3.5 yr in a child with AD, which was confirmed by identification of a novel mutation in FBN1.
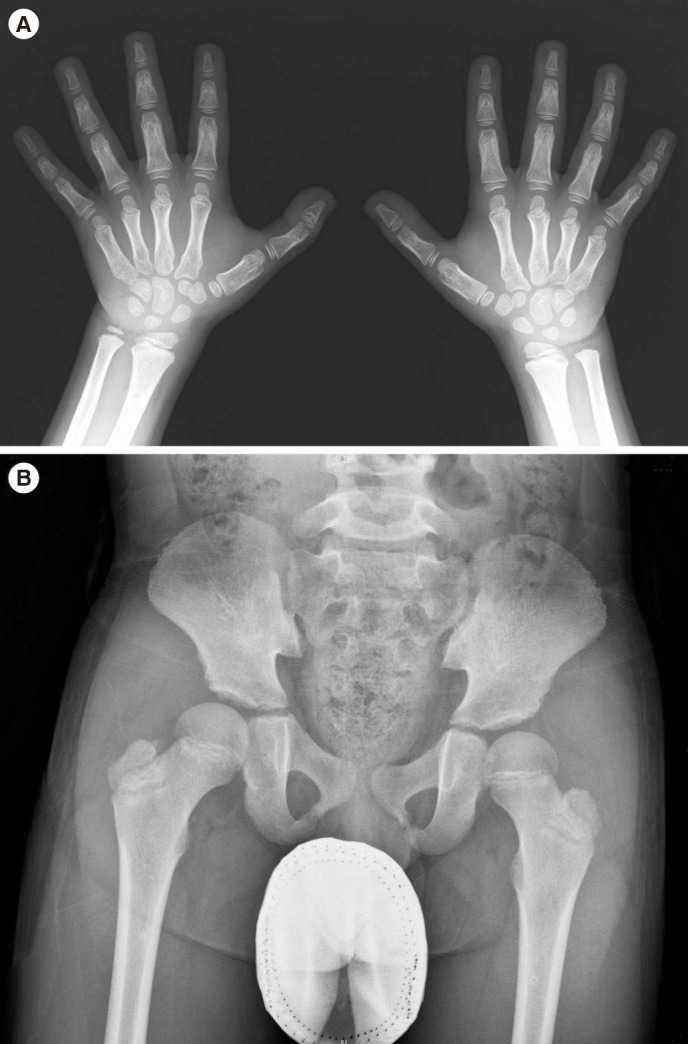
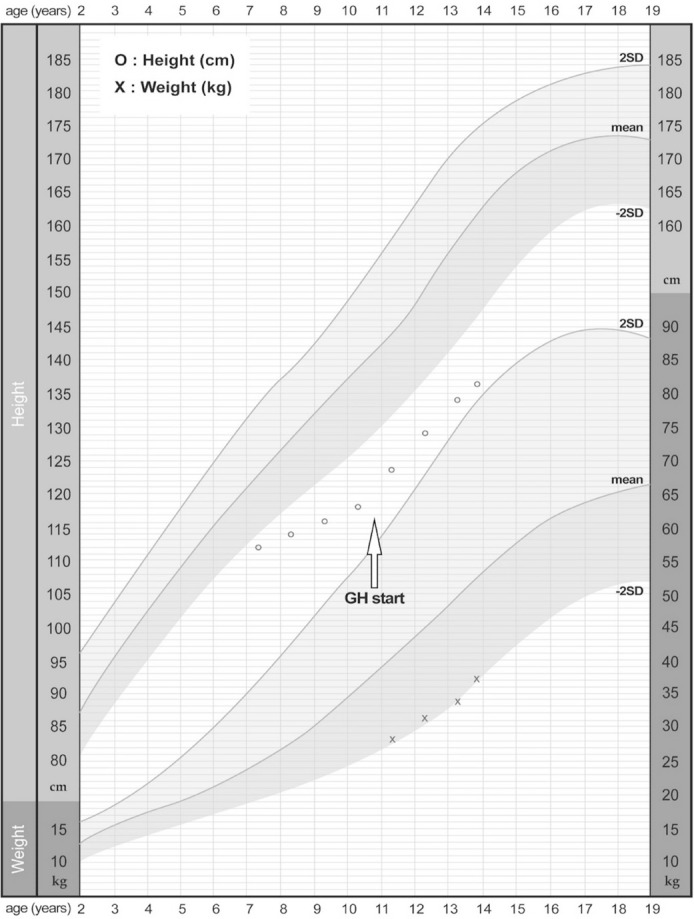
The patient was born to non-consanguineous parents after an uneventful 37 weeks of gestation, with a birth weight of 3,700 g. The patient was referred to our clinic with suspected skeletal dysplasia, at 11 yr of age. He presented short stature, short and stubby hands and feet, normal intelligence, and mild facial dysmorphism: round face, long eyelashes, flat nose with anteverted nostrils, prominent philtrum, and thick lips. He had a bilateral elbow flexion deformity with mild extension limitation. Skeletal survey showed mild brachymetacarpy, delayed bone age (Fig. 1A), and acetabular dysplasia (Fig. 1B). He showed marked growth retardation from 2 yr of age. At 10 yr, his height was 118.1 cm (-3.21 standard deviation score [SDS]); he was diagnosed as having partial growth hormone deficiency at another hospital, and has been treated with rhGH (0.1 IU/kg/day) since then. Subsequently, his growth velocity improved by 5.4 cm/yr, with a height gain of 0.98 SD (Fig. 2). There were no considerable adverse effects during rhGH treatment. Considering the clinical and radiographic findings, AD was the putative diagnosis. We conducted whole-exome sequencing (WES) to simultaneously investigate multiple genes associated with skeletal dysplasia. Written informed consent was obtained from the parents of the patient, and the Institutional Review Board approved this study (IRB file number: 2012-05-080-006). Genomic DNA was extracted from peripheral blood, by using TruSeq Exome Library Prep Kit (Illumina, San Diego, CA, USA) according to the manufacturer's protocols. On the basis of the previously generated BAM file, variants were called using Samtools, smatools mpileup, bcftools view, and vcfutils.pl's varFilter with the maximum depth '-D' set to 1,000, and single nucleotide polymorphism (SNP) and short indel candidates were detected. ANNOVAR (ver. November 2011) was used to annotate these variants with dbSNP135 and the 1,000 Genomes Project SNPs. The variants were further annotated with dbSNP135Common in the UCSC database to identify those with allele frequencies ≥1%, as well as those classified SCS (Clinical Significance), CLN (Variant is Clinical), and the OMIM ID from NCBI dbSNP135, using an in-house program. The WES results identified a heterozygous variant, c.5282C>T (p.Thr1761Ile), in the TB5 domain of FBN1 (NM_000138.4), which was validated by Sanger sequencing. The parents did not carry this variant. All reported mutations in AD are clustered in the same region encoding the TB5 domain of FBN1 [234].
This variant is considered pathogenic for the following reasons: First, Thr1761 in FBN1 is evolutionarily well conserved in mammals. Second, in PolyPhen-2 prediction (http://genetics.bwh.harvard.edu/pph2/), p.Thr1761Ile from c.5282C>T was predicted as "probably damaging" with a score of 0.999. Furthermore, this is a de novo variant, as it was not identified in either parent. AD belongs to the group of acromelic dysplasia, including geleophysic dysplasia (GD) and Weill–Marchesani syndrome (WMS) [5]. In contrast to the genetic heterogeneity of GD and WMS [367], AD seems to be caused only by mutations in FBN1. GD is characterized by characteristic "happy" facial features, hepatomegaly, progressive cardiac disease with valvular abnormality, and tracheal stenosis, often leading to death before 5 yr of age [8]. WMS is distinguished from AD by eye anomalies, including lenticular myopia, ectopia lentis, glaucoma, and spherophakia [9]. Our patient did not show hepatomegaly, valvular heart disease, or ocular disease. He presented with a relatively mild clinical course compared with that of GD or WMS.
GH treatment has not been widely applied in patients with skeletal dysplasia, because genetic heterogeneity and/or clinical variability poses a challenge for assessing the treatment efficacy [10]. rhGH treatment has been reported in only one case of AD, but did not show a significant effect on final height [1]. Our patient received rhGH treatment at 0.1 IU/kg/day for 3.5 yr. During the treatment period, his growth velocity was maintained at up to 5 cm/year, and his height SDS after 3.5 yr was -2.23 SDS. However, long-term follow-up is needed to verify the effects of rhGH treatment.
 XML Download
XML Download