

 PDF
PDF ePub
ePub Citation
Citation Print
Print


Dear Editor,
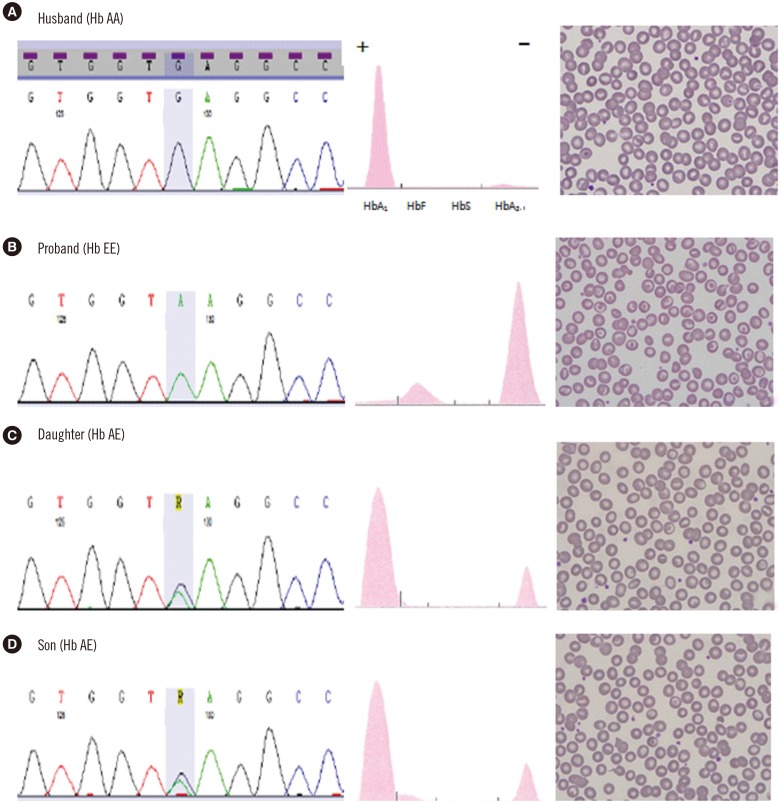
A 33-yr-old Thai woman visited Wonkwang University Sanbon Hospital, Korea, with chief complaints of fatigue and dizziness. Peripheral blood smear examination revealed microcytic hypochromic anemia with target cells (Fig. 1). Routine laboratory analysis results were within normal limits (bilirubin, haptoglobin, reticulocyte, lactate dehydrogenase, iron, total iron binding capacity, ferritin, Coombs' test) except for mild anemia and abnormal Hb electrophoresis (Table 1). Hepatosplenomegaly was absent. Hb electrophoresis was performed on an alkaline agarose gel using a Hydrasis 2 (Sebia, Lisses, France), which revealed markedly increased Hb A2 and Hb F fractions with a severely decreased Hb A1 fraction. Hb E, Hb C, and Hb O migrated at the Hb A2 position during alkaline Hb electrophoresis. To rule out hemoglobinopathy, Homo sapiens Hb β (HBB) (NM_000518.4) gene sequencing was performed. Genomic DNA was extracted from peripheral blood by using a QIAamp DNA mini kit (QIAgen, Hilden, Germany). Two primer sets were utilized that included all exons, splice sites, and adjacent intron regions of the HBB gene. DNA was amplified by using a DNA Engine Tetrad 2 Peltier Thermal Cycler (BIO-RAD, Hercules, CA, USA) and sequenced by using an ABI PRISM 3730XL analyzer (Applied Biosystems, Foster City, CA, USA). Sequencing data were analyzed by using Variant Reporter Software Version 1.1 (Applied Biosystems). The proband's sequencing data revealed a homozygous c.79G>A; p.Glu27Lys (Hb E) variant. A family study revealed Hb E heterozygosity in the proband's two children (Fig. 1).
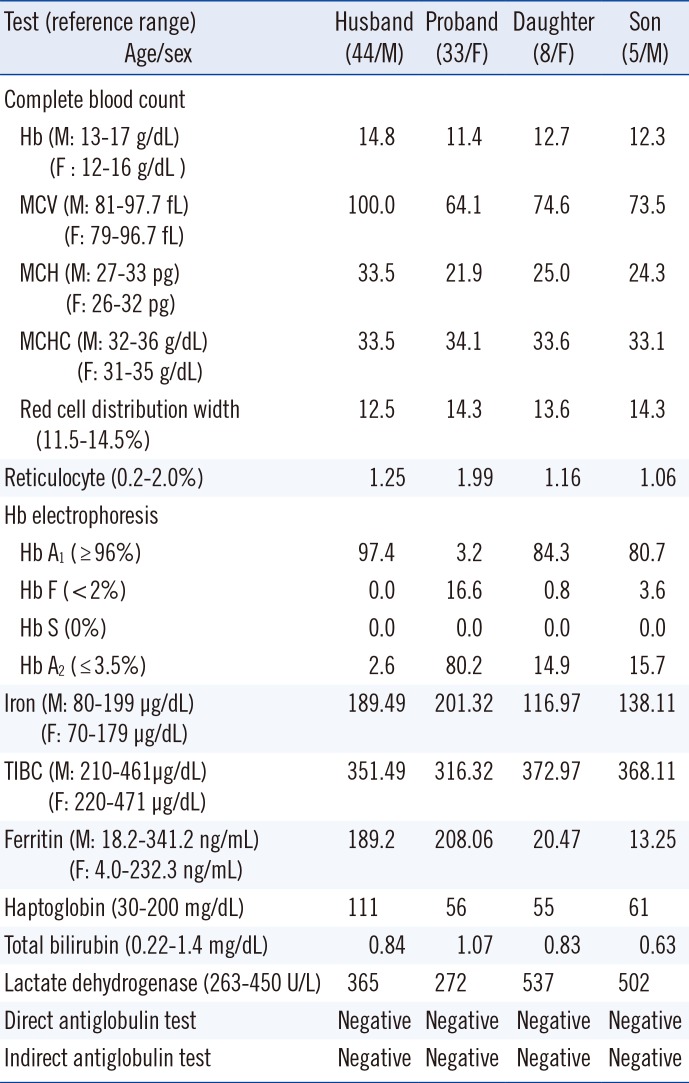
 | Fig. 1Sanger sequencing of the HBB gene, alkaline Hb electrophoresis (see Table 1) and peripheral blood smear (Wright stain, ×1,000) in the Hb E family. (A) Normal findings. (B) Homozygous Hb E (c.79G>A; p.Glu27Lys) variant, increased Hb F and Hb A2 fractions, and many target cells. (C) Heterozygous Hb E, increased Hb A2 and some target cells. (D) Heterozygous Hb E, increased Hb F and Hb A2, and some target cells.
|
Table 1
Laboratory results of the proband's family
![]()
Hemoglobinopathies include all genetic Hb diseases, and are divided into the thalassemia and structural Hb variant groups, which contain many subtypes and combined types. The prevalence of hemoglobinopathies depends on geographic location and ethnicity. Around 7% of the world's population comprises hemoglobinopathy gene carriers. Hb E is the most prevalent variant in Southeast Asia (Thailand, Myanmar, Cambodia, Laos, Vietnam), where its prevalence is 30-60% [1234]. This common structural Hb variant results from the substitution of glutamic acid with lysine at codon 26 of the β globin chain. Hb E disorders may be heterozygous (AE), homozygous (EE), or compound heterozygous (e.g., Hb E with another Hb variant or thalassemia). Hb E is usually associated with a normal Hb concentration or mild anemia, but may also be associated with microcytosis, which may be confused with iron deficiency anemia until laboratory studies are completed. Although Hb E alone does not cause any significant clinical problems, its interactions with other hemoglobinopathies produce a wide range of clinical syndromes. Co-inheritance of Hb E and β-thalassemia, Hb E/β-thalassemia, results in a heterogeneous disease with a phenotype ranging from mild anemia to severe β-thalassemia. To our knowledge, Hb E has not been previously reported in Korea [56]. However, international marriages with Southeast Asians and immigrants importantly contribute to the prevalence of hemoglobinopathy in Korea. This study indicates that clinicians should consider the possibility of hemoglobinopathy in microcytic anemia.
 XML Download
XML Download